










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Farmacia
Print version ISSN 0034-7515
Rev Cubana Farm vol.47 no.3 Ciudad de la Habana Jul.-Sept. 2013
ARTÍCULO ORIGINAL
Evaluación de métodos cromatográficos para la estabilidad química del nitrato de miconazol en una nueva crema
Assessment of chromatographic methods for the chemical stability of a new miconazol nitrate cream
MSc. Oscar García Pulpeiro,I Lic. Wendy Calzadilla Aguiar,II Lic. Wendy Rodríguez Bencomo,II Lic. Eduardo Rodolfo Besada Maribona,I Dra. C. Yania Suárez PérezII
I Empresa Laboratorio "Roberto Escudero Díaz". La Habana, Cuba.
II Instituto de Farmacia y Alimentos. Universidad de La Habana. La Habana, Cuba.
RESUMEN
Objetivo: evaluar los métodos cromatográficos para la estabilidad química del nitrato de miconazol en una nueva crema al 2 %.
Métodos: en primer lugar se aplicaron diferentes condiciones degradativas al nitrato de miconazol materia prima a fin de obtener los posibles productos de degradación del fármaco y evaluarlos por un método diseñado por cromatografía en capa delgada, el cual se validó para identificar productos de degradación en la crema. Se evaluó el desempeño del método oficial informado en la Farmacopea Británica 2010 por cromatografía líquida de alta resolución para la valoración del nitrato de miconazol en la crema, analizando su selectividad frente a los posibles productos de degradación. Ambos métodos cromatográficos fueron aplicados al análisis de muestras de crema procedentes de los tres lotes pilotos sometidos a estrés térmico durante 30 días.
Resultados: ambos métodos mostraron elevada selectividad frente a los excipientes y los productos de degradación del fármaco. Se obtuvo degradación del nitrato de miconazol frente a hidrólisis ácida, termólisis y fotólisis y el límite de detección fue de 1 µg para cromatografía en capa delgada. No se mostró degradación del analito según los resultados cualitativos y cuantitativos en ninguno de los tres lotes analizados.
Conclusiones: los métodos utilizados son válidos para el objetivo con el cual se proponen, por lo que pueden emplearse en el estudio de estabilidad química de las cremas de nitrato de miconazol al 2 %.
Palabras clave: nitrato de miconazol, crema, cromatografía en capa delgada, cromatografía líquida de alta resolución, validación.
ABSTRACT
Objective: to assess the chromatographic methods for the chemical stability of a new 2 % miconazol nitrate cream.
Methods: various degradation conditions were firstly used in the raw material miconazole nitrate in order to obtain the possible degradation products of this drug and to evaluate them by thin layer chromatography-based method, which was validated to identify the degradation products in the new cream. The performance of the official method based on high resolution liquid chromatography and reported in British Pharmacopeia 2010 was evaluated, and its selectivity against the possible degradation products were also analyzed. Both chromatographic methods were applied to the analysis of cream samples from the three pilot batches under heat stress for 30 days.
Results: the two methods showed high selectivity against excipients and degradation products of the drug. Miconazol nitrate was degraded against acid hydrolysis, thermolysis and photolysis, being the detection limit of 1 µg for the thin layer chromatography. No degradation of the analyte was observed in any of the three analyzed batches according to the qualitative and quantitative results.
Conclusions: these methods are valid for the submitted objective, so they may be used in the chemical stability study of 2 % miconazol nitrate creams.
Key words: miconazol nitrate, cream, thin layer chromatography, high performance liquid chromatography, validation.
INTRODUCCIÓN
El nitrato de miconazol tiene un amplio espectro como antifúngico. Es un compuesto de tipo imidazólico con limitada solubilidad en agua, a pesar de encontrarse en forma de sal. Se utiliza fundamentalmente por vía tópica en micosis superficiales, como es el caso de infecciones dermatofíticas del tipo tinea pedis, tinea cruris, tinea versicolor, así como candidiasis cutánea y vaginal.1
En Cuba se comercializa en forma de crema al 2 %. Recientemente se llevó a cabo la reformulación de este producto, teniendo en cuenta desviaciones presentadas respecto al cumplimiento de algunas especificaciones de calidad física y microbiológica.2 Como resultado se realizaron importantes cambios en la composición de la formulación. Como parte de los estudios exigidos para registrar la crema reformulada, se encuentra el desarrollo de métodos analíticos que permitan conocer el comportamiento del fármaco en el tiempo a través de los estudios de estabilidad química, por lo que constituye un aspecto clave, la validación de estos.3
Los métodos cromatográficos son técnicas indicadoras de estabilidad que constituyen métodos de elección para el seguimiento de la estabilidad química de los fármacos.4 Para el nitrato de miconazol no se han informado métodos cromatográficos con estos fines, sin embargo, se utilizan para la cuantificación en diversas formas farmacéuticas. Tal es el caso del uso de cromatografía gaseosa para productos semisólidos. Se recomienda para su cuantificación en un gel oral usando flurazepam 60 mg mL-1 como estándar interno y detección nitrógeno-fosfórica.5 Para análisis de óvulos se obtuvo un método sensible con límite de detección de 3,1 µg mL-1 empleando detector de ionización de llama.6
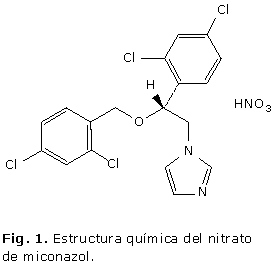
La cromatografía líquida de alta resolución (CLAR) es el método oficial para la cuantificación del nitrato de miconazol en la crema, empleando un sistema isocrático a 45 °C y una velocidad de flujo de 1 mL min-1 con el uso de ácido benzoico como estándar interno7 o utilizando una solución de nitrato de econazol en mezcla de volúmenes iguales de metanol y tetrahidrofurano con detección a 235 nm y 2 mL min-1 como velocidad de flujo.8 El empleo de detector UV7,8 se justifica teniendo en cuenta la presencia de grupos cromóforos en la estructura del compuesto (Fig. 1).
Además se reporta el uso de cromatografía líquida de ultra desempeño (UHPLC) con columnas cromatográficas sub-2 µm como método de determinación cuantitativa de miconazol y sus sustancias relacionadas, en aras de reducir el tiempo de análisis y el consumo de disolvente en la cromatografía líquida.9
La validación exhaustiva de estos métodos, o en su defecto, la evaluación del desempeño según el objetivo propuesto para su uso, garantizan la comprobación confiable y reproducible de los índices de calidad requeridos.3
Teniendo en cuenta estos antecedentes, se propone como objetivo evaluar los métodos cromatográficos para la estabilidad química del nitrato de miconazol en una nueva crema al 2 %. Para ello se desarrolló y validó un método por cromatografía en capa delgada (CCD) para la identificación de los posibles productos de degradación del nitrato de miconazol en la crema recién reformulada y se evaluó el desempeño del método por CLAR informado en la Farmacopea Británica (BP) 2010 para el análisis del nitrato de miconazol en la crema.
MÉTODOS
Obtención de los posibles productos de degradación del nitrato de miconazol
Se sometió el ingrediente farmacéutico activo (IFA) de nitrato de miconazol (GI: 541007020), a condiciones de estrés con el objetivo de obtener los posibles productos de degradación según las condiciones recomendadas por ICH Q1A (R2), 2003.10
Las condiciones degradativas aplicadas fueron las siguientes:
- Termólisis: se colocó una porción de 1 g de nitrato de miconazol (IFA) en una cápsula de porcelana y se sometió a calentamiento a 100 ± 5 °C durante 1 h en la estufa P Selecta (España).
- Hidrólisis neutra: se disolvió 1 g de IFA en 50 mL de metanol y se añadieron 50 mL de agua destilada. La muestra se sometió a reflujo durante 1 h.
- Hidrólisis en medio ácido: se disolvió 1 g de IFA en 50 mL de metanol y se añadieron 50 mL de HCl 1 mol/L. La muestra se sometió a reflujo durante 4 h.
- Hidrólisis en medio básico: se disolvió 1 g de IFA en 50 mL de metanol y se añadieron 50 mL de NaOH 1 mol/L. La muestra se sometió a reflujo durante 1 h.
- Oxidación: se disolvió 1 g de IFA en 50 mL de metanol y se trasvasó a un vaso de precipitado de capacidad 100 mL. Se añadieron 10 mL de agua destilada y 3 gotas de H2O2. La muestra se colocó en baño de María durante 1 h.
- Luz natural: se disolvió 1 g de IFA en 50 L de metanol y se añadieron 50 mL de agua destilada. La muestra se colocó en un tubo de ensayo de cristal transparente con tapa y se expuso a la luz solar directamente durante 7 días.
Para las muestras sometidas a reflujo, se empleó un balón de capacidad 250 mL previamente tarado, un condensador de bolas de boca esmerilada y mangueras de silicona. La temperatura fue suministrada por una plancha eléctrica. Finalizado el reflujo, las disoluciones resultantes una vez alcanzada la temperatura ambiente, se verificaron por pesada para comprobar que no se había perdido muestra durante el procesamiento aplicado. Se utilizó la balanza analítica Sartorius AG, modelo BS124S (China).
Se preparó una solución de referencia (SR1) de nitrato de miconazol, para lo cual se pesó 1 g de IFA y se disolvió en 100 mL de metanol, para obtener una concentración final de 0,01 % (m/v).
De la muestra sometida a termólisis se toma la porción necesaria para lograr una disolución en metanol de igual concentración que la solución SR1.
Todas las muestras sometidas a condiciones de estrés se trasvasaron a vasos de precipitado. Las soluciones se concentraron a 60 ± 0,5 °C en baño de agua termostatado para eliminar aproximadamente la tercera parte del volumen total. Posteriormente se trasvasaron a embudos separadores. Se añadió NaOH 1 mol/L hasta alcalinizar las muestras a pH³ 8 (para lo cual se determinó el volumen necesario a añadir previamente con ayuda de papel indicador de pH) y 30 mL de cloroformo. Los extractos clorofórmicos resultantes de una extracción fueron recolectados para emplearlos posteriormente en la evaluación por CCD.
Método por CCD para estudios de estabilidad del nitrato de miconazol en la crema al 2 %. Condiciones cromatográficas
Las condiciones cromatográficas utilizadas fueron las siguientes:
- Fase estacionaria: placas sin activar de sílica gel GF254.
- Fase móvil: éter de petróleo: cloroformo:metanol:solución amoniacal al 25 % (60:30:10:1 v/v/v/v).
- Revelador: luz UV suministrada por una lámpara UV HANAU (China) a 254 nm.
Se preparó una solución de referencia (SR2) de nitrato de miconazol a una concentración de 1,25 mg/mL en metanol. Se realizaron las corridas cromatográficas en cámaras previamente saturadas. De cada muestra resultante del procedimiento degradativo aplicado, se puntearon 10 mL. Para cada mancha obtenida al realizar el revelado, se determinó el Factor de retardo (Rf).
Criterio de aceptación: el método se consideró selectivo si se observa la mancha correspondiente al nitrato de miconazol (SR) bien definida, separada del punto de aplicación y del frente de disolvente y con Rf diferente a cualquier otra mancha obtenida (productos de degradación).
SR1 se utilizó para el ensayo de selectividad para estabilidad y SR2 se empleó para el análisis de las cremas degradadas
Procedimiento para la aplicación del método por CCD al análisis de la crema
Teniendo en cuenta que el método por CCD se propone para el análisis de la crema, se diseñó un procedimiento para la toma de la muestra desde la forma farmacéutica, el cual se describe a continuación:
Se pesó la porción de crema equivalente a 0,05 g de IFA en un vaso de precipitado de capacidad 150 mL y se añadieron 5 mL de agua. Se dispersó la crema con ayuda de agitación. Posteriormente se trasvasó a un embudo separador y se añadieron 5 mL de NaOH 1 mol/L hasta alcalinizar el medio, logrando un valor de pH³ 8. Se realizaron dos extracciones con 15 mL de cloroformo, agitando vigorosamente y se dejaron separar las fases. Se colectaron las fases clorofórmicas y se filtraron sobre Na2SO4 anhidro. Se tomó una alícuota de 10 µL del filtrado y se aplicó en la placa cromatográfica.
Se realizó el corrimiento cromatográfico y se reveló con luz UV. Finalmente se midió el Rf de cada mancha obtenida respecto a la SR2.
Criterio de aceptación: la mancha correspondiente al nitrato de miconazol en la muestra debe coincidir en localización (Rf) con la mancha obtenida para las SR2, libre de interferencia de manchas secundarias.
Validación del método por CCD para estudios de estabilidad del nitrato de miconazol en la crema
Se tuvieron en cuenta los criterios recomendados para los métodos de la categoría II cualitativa.7
- Evaluación de la selectividad: Se puntearon 10 µL de una SR de nitrato de miconazol en metanol equivalente al 100 % (SR1) y de cada una de las muestras degradadas obtenidas en cloroformo, una vez aplicadas diferentes condiciones degradativas descritas con anterioridad.
Para considerar la influencia de los restantes componentes de la formulación, se prepararon placebos de la formulación propuesta.2 En otra placa se puntearon 10 µL de la solución clorofórmica resultante de procesar placebos del producto según el procedimiento descrito para el análisis de las cremas y se compararon con la respuesta obtenida con la SR2. También se aplicaron 10 µL de placebos degradados térmicamente en una estufa P Selecta ajustada a 45 ± 5 °C durante 7 días. En cada caso se determinó el factor de retardo (Rf) y se verificó la capacidad del método para detectar la presencia del IFA libre de interferencia de los componentes de la matriz y de los posibles productos de degradación del fármaco y de los excipientes (selectividad para estabilidad).
- Evaluación del límite de detección: Se preparó una solución de nitrato de miconazol a concentración de 1 mg/mL en metanol. Se puntearon en una misma placa volúmenes de 20, 10, 5, 2 y 1 µL respectivamente y se realizó el corrimiento cromatográfico y el revelado de la placa bajo luz UV. Se determinó la mínima cantidad de analito detectada en estas condiciones, lo cual definió la selección del límite de detección (Ld).
Método por CLAR para nitrato de miconazol en la crema
Se utilizó el mismo método descrito en la BP 20108 para la cuantificación del nitrato de miconazol en la crema, el cual se describe a continuación:
Preparación de las muestras y patrones:
- Preparación del patrón de nitrato de miconazol: se preparó una solución al 0,1 % m/v de nitrato de miconazol SR USP en una mezcla de iguales volúmenes de metanol y tetrahidrofurano.
- Preparación de la muestra: se pesó la cantidad de crema equivalente a 50 mg de nitrato de miconazol y se agita con 30 mL de una mezcla de iguales volúmenes de metanol y tetrahidrofurano durante 30 min. Posteriormente se añade suficiente mezcla de disolvente hasta completar volumen con exactitud empleando un matraz aforado de capacidad 50 mL y se filtra la disolución resultante por un filtro Milipore de teflón 0,45 µm (Merck).
Condiciones cromatográficas: las condiciones cromatográficas fueron las mismas reportadas para la determinación de sustancias relacionadas en la BP 20108 y fueron las siguientes:
- Equipo: Smart Line KNauer
- Fase estacionaria: columna MEDITERRANEA SEA RP C18 5µ m (10 x 0,21).
- Fase móvil: solución de acetato de amonio 0,6 % m/v en una mezcla de 300 mL de acetonitrilo, 320 mL de metanol y 380 mL de agua.
- Velocidad de flujo: 2 mL/min.
- Detector UV-VIS KNauer: 235 nm.
Prueba de adecuación del sistema: Se determinó la resolución (Rs) entre el nitrato de econazol y el nitrato de miconazol sustancias de referencia de calidad USP. Ambas soluciones se prepararon en metanol en concentraciones de 0,0025 % (m/v) y se mezclaron en volúmenes iguales de metanol y tetrahidrofurano.
Criterio de aceptación: Rs³ 1,5.
Evaluación del desempeño del método por CLAR para estudios de estabilidad del nitrato de miconazol en la crema
Se procedió a evaluar la capacidad del método para dar respuesta con el nitrato de miconazol libre de interferencia de los restantes componentes de la formulación. Para ello se inyectaron en el cromatógrafo 20 µL de las muestras de placebo procesadas de igual forma que la descrita para las cremas. Además se inyectaron iguales volúmenes de los productos de degradación obtenidos, los cuales fueron disueltos en una mezcla de metanol:tetrahidrofurano (1:1) y se filtraron antes de inyectar al equipo (selectividad para estabilidad).
El método se consideró válido para su aplicación a los estudios de estabilidad de la crema al 2 % de nitrato de miconazol, si no se detectan señales secundarias con igual tr al del nitrato de miconazol SR USP. Los resultados obtenidos se procesaron a través del Software Chrom Gate.
Aplicación de los métodos analíticos propuestos para estudios de estabilidad al análisis de muestras de cremas
Se elaboraron tres lotes de 5 kg de cremas de la formulación seleccionada2 rotulados como lotes EP 11001, EP 11002 y EP 11003, usando un método muy similar al empleado a escala de laboratorio.2 Se realizaron análisis por triplicado a muestras de crema de cada lote, utilizando muestras colocadas en estufa ajustada a 45 ± 5 °C durante 30 días. En cada lote se aplicó el análisis cuantitativo (método por CLAR) y el análisis cualitativo (método por CCD).
RESULTADOS
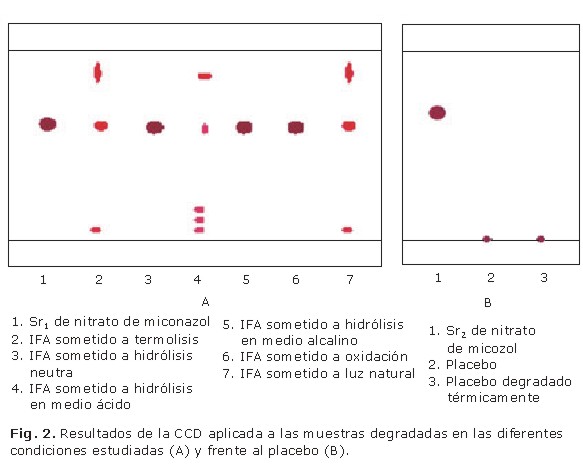
En la figura 2 (A) se reflejan los resultados obtenidos en la CCD aplicada a las muestras degradadas en las diferentes condiciones estudiadas para la degradación del IFA.
Como se observa, se obtuvo degradación del nitrato de miconazol por efecto de la temperatura (2), la hidrólisis catalizada en medio ácido (4) y la luz (7). Los resultados obtenidos para el efecto de termólisis y luz natural fueron muy similares, ya que se detectaron dos manchas adicionales a la del IFA, una muy próxima al punto de aplicación (Rf= 0,04) y otra que se aproximó al frente del disolvente (Rf= 0,77). Sin embargo, la hidrólisis en medio ácido provocó la aparición de cuatro posibles productos de degradación del nitrato de miconazol y una apreciable disminución en la intensidad de la mancha con Rf similar al IFA. De las cuatro manchas secundarias, dos mostraron iguales valores de Rf a las manchas secundarias obtenidas en termólisis y luz natural; lo cual sugiere que estas se corresponden con los productos de degradación más probables.
La buena resolución alcanzada entre IFA (Rf= 0,60) y sus posibles productos de degradación (Rf de 0,04; 0,07; 0,10; 0,77) sugiere que las condiciones cromatográficas seleccionadas fueron adecuadas para el objetivo con el cual se propone este método.
En la figura 2 (B) se representan los resultados de la selectividad del método frente a los placebos. Solo se obtuvo una mancha muy pequeña en el punto de aplicación. Tampoco se observaron posibles productos de degradación de los excipientes.
El Ld fue 1 µg, ya que esta fue la menor concentración de analito que se visualizó en la placa con adecuada intensidad. Corresponde con el menor volumen aplicado (1 µL) en la CCD.
El conjunto de estos resultados permite afirmar que este método fue válido para el seguimiento del nitrato de miconazol en la crema al 2 % durante los estudios de estabilidad, como método cualitativo usado simultáneamente con un método cuantitativo.
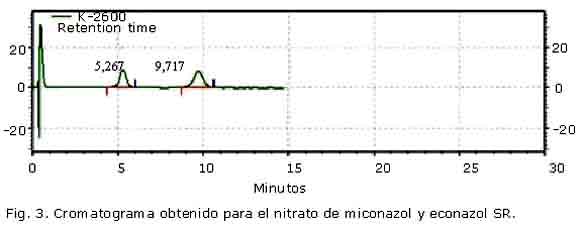
En la figura 3 se muestra el cromatograma obtenido para las SR de miconazol y econazol. Se observa una adecuada resolución entre las señales obtenidas, ya que Rs fue de 2,405; valor que supera el límite establecido; lo cual constituye un aval de la adecuación del sistema cromatográfico empleado. La respuesta para el miconazol se obtuvo a un tr= 9,72; mientras que la del econazol aparece a un menor tiempo de retención (tr= 5,26).
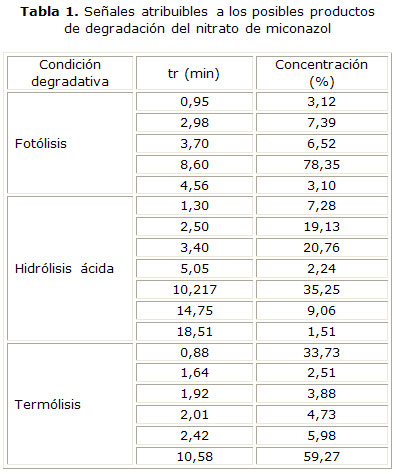
Los resultados del ensayo de selectividad se muestran en la figura 4. No se detectaron señales analíticas correspondientes a los excipientes, por lo que se comprobó que el cambio de matriz no afectó la selectividad del método para control de calidad. La selectividad del método frente a los productos de degradación del IFA se muestra en la figura 4 (B-G). El pico principal en todos los casos fue atribuible al nitrato de miconazol, con un tr similar al obtenido para la SR. Las señales atribuibles a los posibles productos de degradación del IFA mostraron tr diferentes al analito y al placebo, por lo que el método fue suficientemente selectivo para aplicarlo a estudios de estabilidad química del nitrato de miconazol en la crema.
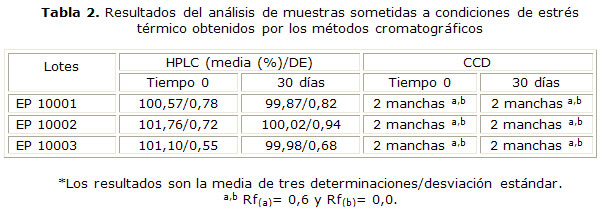
Los resultados obtenidos con este método, en general coincidieron con los obtenidos por CCD, ya que las condiciones en las cuales se obtuvieron las principales señales atribuibles a los productos de degradación fueron la hidrólisis ácida (Fig. 4) (E), seguida de termólisis (Fig. 4) (F) y fotólisis (Fig. 4) (D). Aunque en la hidrólisis neutra y alcalina se detectan pequeñas señales a tr inferiores al nitrato de miconazol, la señal del analito fue la mayor (Fig. 4) (B y C), mientras que la oxidación no se detectaron señales cuantificables (Fig. 4) (G) (tabla 1). La tabla 1 resume los tr de todas las señales analíticas cuantificables, para la condición degradativa: fotólisis, hidrólisis ácida y termólisis. Aparece sombreado en cada caso la señal atribuible al IFA. En la tabla 2 se presentan los resultados cuantitativos y cualitativos derivados de la aplicación de los métodos cromatográficos al análisis de muestras degradadas en condiciones de estrés térmico por un período de 30 días. Los resultados mostrados son la media de 3 determinaciones. Las respuestas analíticas de las muestras de crema procedentes de los tres lotes analizados, tuvieron el mismo comportamiento.
DISCUSIÓN
En primer lugar fue necesario degradar el IFA empleando condiciones de estrés para conocer las principales vías degradativas que afectan al nitrato de miconazol, ya que no hubo informes relacionados con este tema en la bibliografía consultada. Solo se encontraron referencias relacionadas con la "protección de la luz" propuestas por las Farmacopeas.7,8
Los estudios de estrés permiten dilucidar la estabilidad intrínseca de un analito y forman parte de la estrategia a seguir cuando los productos de degradación de un fármaco no son conocidos. A su vez, son de un valor inestimable para la validación de los métodos analíticos y emplean condiciones más severas que las pruebas de estabilidad acelerada. Se seleccionaron las condiciones degradativas sugeridas por la Guía ICH Q1A (R2):10 temperatura, hidrólisis en diferentes rangos de pH, medio oxidante y luz. Para la evaluación del efecto de la luz no se contaba con los aditamentos requeridos para los estudios de fotosensibilidad,11 por lo que se utilizó la luz natural. Solo se omitió la evaluación del efecto de la humedad, ya que se trata de una crema y esta condición se analizará en los estudios de estabilidad definitivos.
La posibilidad de obtención de cuatro productos de degradación en medio ácido (Fig. 2) (A), se justifica teniendo en cuenta la probabilidad de que ocurra ruptura por el enlace éter presente en el nitrato de miconazol en estas condiciones. En dependencia del lugar por donde ocurra esta ruptura (Fig. 1), se obtendrá uno u otro producto de degradación, sin embargo, en medio ácido fue posible obtener las cuatro posibilidades. Esto indicó que las condiciones degradativas aplicadas fueron suficientes para generar los posibles productos de degradación de este fármaco. Las manchas que solo se visualizaron en la muestra sometida a hidrólisis en medio ácido, se localizaron en una zona próxima al punto de aplicación, con intensidades menores que el resto y con valores de Rf de 0,07 y 0,10 respectivamente.
Con relación al procedimiento propuesto para la aplicación del método por CCD al análisis de la crema, se debe destacar la extracción del IFA en forma de base en los extractos clorofórmicos, una vez que se desplaza el equilibrio a la forma no disociada cuando se alcaliniza el medio. La filtración sobre Na2SO4 anhidro garantiza la remoción de cualquier resto de fase acuosa en la fase clorofórmica en la cual se encuentran los analitos de interés para la CCD. El revelado físico con luz UV es un método muy sencillo, se ahorra tiempo y reactivos, si se compara con el revelado químico y resulta una opción muy útil en compuestos como este que presentan cromóforos en su estructura. En este caso el revelado con luz UV, también garantiza la visualización de los posibles productos de degradación, pues independientemente de por donde se produzca la ruptura del enlace éter, los compuestos resultantes también presentan grupos cromóforos (Fig. 1).
La selectividad del método por CCD, se complementó al analizar placebos del producto en las mismas condiciones cromatográficas propuestas para las muestras sometidas a condiciones de estrés, ya que su respuesta analítica no debe interferir con ninguna de las manchas atribuibles al IFA ni a sus productos de degradación. Los resultados de la selectividad frente a placebos sugieren que el procedimiento propuesto para el tratamiento de las muestras fue efectivo (Fig. 2) (B), lo cual confirma que los productos de degradación (manchas secundarias) obtenidas en las muestras sometidas a termólisis se deben a la degradación del IFA y no a la degradación de otros componentes de la crema. Las manchas presentes en el punto de aplicación se atribuyeron a los preservos usados en la formulación (metil y propilparabeno), ya que son los únicos componentes de la formulación capaces de dar respuesta frente a detector UV. Estos resultados confirman la adecuada selectividad del método para el uso previsto.
La menor concentración detectada (Ld= 1 µg) se consideró muy baja con respecto a las concentraciones de nitrato de miconazol presentes en la crema cuando los niveles de degradación alcancen el 90 % de la concentración inicial, por lo que el método fue suficientemente sensible para el objetivo con el cual se propone.
Los métodos cromatográficos instrumentales son técnicas indicadoras de estabilidad, que permiten diferenciar y al mismo tiempo cuantificar, la concentración de analito libre de interferencias de los principales productos de degradación que pueden estar presentes durante la conservación de un producto farmacéutico.4
El método informado en la BP 20108 se basa en una separación por reparto en fase reversa, donde la fase estacionaria es apolar y la fase móvil es medianamente polar. La detección se realiza a l= 235 nm, teniendo en cuenta la presencia de cromóforos en la estructura del compuesto (Fig. 1), el cual muestra respuesta en el espectro UV a partir de los 230 nm. Un método oficial que se aplica con el mismo propósito con el que aparece informado en la Farmacopea, no requiere validación.3 Sin embargo, cuando se trata de una forma farmacéutica, cuya composición es variable, es preciso evaluar al menos parcialmente, la validez del método analítico en cuestión. Esta validación parcial, tiene como principal objetivo, comprobar el impacto de los componentes de la formulación en los resultados analíticos. Si adicionalmente, se modifica el alcance del método oficial, se hace aun más justificada la evaluación del desempeño del método.3
Este es el caso del método por CLAR que se propone en este trabajo, ya que se informa para control de calidad en la BP 20108 y se propone para el seguimiento de la estabilidad química del IFA en la crema durante los estudios de estabilidad, requeridos como etapa indispensable para lograr la modificación del registro por parte de la Autoridad Reguladora a nivel nacional.12
El cambio de matriz y de alcance en el método, justifica la evaluación del desempeño de la técnica cromatográfica oficial. En este método, el parámetro de validación más importante es la selectividad que demuestra la capacidad del mismo de dar respuesta libre de interferencia de los restantes componentes de la formulación (selectividad para control de calidad) y de los posibles productos de degradación del IFA y de los excipientes (selectividad para estabilidad). La garantía de que los restantes componentes de la formulación no afectan el resultado analítico, confirma que el cambio en la matriz no afecta los resultados obtenidos. Por otra parte, se debe considerar que no se realizó ningún cambio en el procedimiento normalizado que aparece en la BP 20108 para al análisis de las cremas, por lo que el impacto en parámetros como precisión y exactitud no fueron de interés en este trabajo. Teniendo en cuenta la modificación del alcance de control de calidad (método oficial) a estudios de estabilidad (uso propuesto), resultó imprescindible retar al método frente a los posibles productos de degradación que eventualmente, pudieran coexistir con el analito en la matriz durante los estudios de estabilidad química.
La variabilidad observada en el tr del nitrato de miconazol se atribuye a variaciones en las condiciones experimentales durante la ejecución de los ensayos, por ejemplo, días y columnas diferentes.
La degradación en medio ácido superó el 50 %, lo que muestra la influencia de este medio en la estabilidad química del nitrato de miconazol. La degradación por efecto de la temperatura fue próxima al 40 % de la concentración inicial, mientras que el efecto de la luz degradó aproximadamente un 20 % del nitrato de miconazol presente en la muestra.
El sistema cromatográfico propuesto en el método oficial de la BP 2010,8 resultó válido para cuantificar el nitrato de miconazol en la crema libre de interferencias de los posibles productos de degradación del IFA. Por esta razón se puede sugerir la aplicación de este método al seguimiento de la estabilidad química de los lotes pilotos de la crema al 2 % reformulada2 en aras de mejorar su calidad.
Transcurrido un mes de almacenadas las muestras a 45 ± 0,5 °C, solo se obtuvieron picos atribuibles al IFA en la CLAR, que representaron concentraciones muy próximas al 100 % de analito, en cada lote analizado. En correspondencia con los resultados cuantitativos, la CCD solo permitió detectar manchas de igual Rf (Rf= 0,6) e intensidad que el IFA presente en la SR2 y la correspondiente a los parabenos presentes en la formulación en el punto de aplicación (Rf= 0).
Los métodos por CCD y por CLAR resultan válidos para el objetivo con el cual se proponen, por lo que pueden emplearse en el estudio de estabilidad química de las cremas de nitrato de miconazol al 2 %.
REFERENCIAS BIBLIOGRÁFICAS
1. Mediavilla A, Flórez J. Fármacos antifúngicos. Farmacología humana. 3ra. ed. Barcelona: Masson, SA; 1997. p. 118.
2. Rodríguez W. Reformulación de la crema de nitrato de miconazol al 2 % [tesis]. La Habana: Instituto de Farmacia y Alimentos. Universidad de La Habana; 2012.
3. Centro para el Control Estatal de la Calidad de los Medicamentos. Regulación 41. Validación de métodos analíticos. La Habana: CECMED; 2007.
4. Castiñeira M, González HM. Control de Medicamentos 2da. ed. La Habana: Félix Varela; 2002. p. 23.
5. Ros JW, Van der Meer YG. Preparation of Miconazole Oral Gel. Pharm Weekbl. 1990;125:70-1.
6. Ashour S, Kattan N. Determinación simultánea de nitrato de miconazol y metronidazol en diferentes formas farmacéuticas por cromatografía de gases y detector de ionización de llama (GC-FID). Rev Intern de Cienc Bioméd. 2010;6(1):13-8.
7. The United Stated Pharmacopoeia 33 and National Formulary 28 (USP33-NF28). Validation of compendial procedures. 33 ed. Rockville: Mack Printing; 2010.
8. British Pharmacopoeia. Volume I & II. Monographs: Medicinal and Pharmaceutical Substances-Miconazole Nitrate. London: The Stationery Office; 2010. Versión electrónica 14.0.
9. O'Connor N; Geary, M. The Determination of Miconazole and its Related Production Impurities Together with Basic Solution Stability Studies Using a Sub 2 µm Chromatographic Column. J Cromatogr Sci. 2012;50(3):199-205.
10. ICH Q1A (R2). Harmonised Tripartite Guideline. Stability Testing of new drug substances and products (Stress Testing). Geneva: ICH; 2003.
11. ICH Q1B. Harmonised Tripartite Guideline. Photostability Testing of New Drug Substances and Products. Geneva: ICH; 1996.
12. Centro para el Control Estatal de la Calidad de los Medicamentos. Regulación 23: Requerimientos para los estudios de estabilidad para el registro de productos farmacéuticos nuevos y conocidos. La Habana: CECMED; 2000.
Recibido: 4 de marzo de 2013.
Aprobado: 6 de mayo de 2013.
Yania Suárez Pérez. Instituto de Farmacia y Alimentos. Universidad de La Habana. Ave 23 No. 21425 e/ 214 y 222, La Coronela, La Lisa, CP 13600, La Habana, Cuba. Correo electrónico: yaniasp@ifal.uh.cu, yania_as@yahoo.es


{kind=link}
{kind=link}
{kind=link}