










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Farmacia
Print version ISSN 0034-7515
Rev Cubana Farm vol.48 no.1 Ciudad de la Habana Jan.-Mar. 2014
ARTÍCULO ORIGINAL
Validación de un método de análisis para la determinación de raloxifeno en una formulación cubana
Validation of an analysis method to determine raloxifene in a Cuban formulation
DrC. Jorge E. Rodríguez Chanfrau, Téc. Marilyn López Armas
Centro de Investigaciones y Desarrollo de Medicamentos (CIDEM). La Habana, Cuba.
RESUMEN
Introducción: el raloxifeno es un modulador selectivo del receptor estrogénico, perteneciente a la familia de los benzotiofenos. Los resultados de varios ensayos clínicos han mostrado que este fármaco reduce la pérdida del hueso en la espina dorsal y puede aumentar la masa del hueso en ciertos sitios.
Objetivo: validar un método de análisis para la determinación de raloxifeno en una formulación cubana.
Métodos: se desarrolló un método de análisis por cromatografía líquida de alta resolución. Se emplearon para ello una columna Lichrospher® 100, RP 18. Se utilizaron como fase móvil una mezcla de metanol-agua desionizada-trietanolamina (70:30:0,1) y detector ultravioleta a una longitud de onda de 286 nm. El flujo de la fase móvil fue de 1,5 mL/min y el volumen de inyección fue de 20 mL.
Resultados: el método empleado para la cuantificación de raloxifeno en la formulación cubana resultó específico, preciso, exacto y lineal.
Conclusiones: en las condiciones de estudio se demuestra que el método propuesto cumple con los parámetros de validación, por lo que puede ser utilizado en el control de la calidad de dicho medicamento.
Palabras clave: raloxifeno, método de análisis, cromatografía líquida de alta resolución, validación.
ABSTRACT
Introduction: raloxifene is a selective estrogen receptor modulator from the benzothiophene family. The results of several large clinical trials have shown that raloxifene reduces the rate of bone loss in the dorsal spine and may increase bone mass at certain sites.
Objective: to validate an analytical method for determination of raloxifene in a Cuban formulation.
Methods: a high performance liquid chromatography analytical method has been developed for the estimation of raloxifene in Cuban pharmaceutical dosage form. In this method, RP 18 Lichrospher® 100 column with mobile phase consisting of methanol, de-ionized water and trietanolamine (ratio of 70:30:0.1) and 286 nm wavelength ultraviolet detector were used. The mobile phase flow was 1.5 mL/min and the injected volume reached 20 mL
Results: the results showed that the suggested method for raloxifene quantitation was specific, accurate, precise and linear.
Conclusions: under the studied conditions the analytic procedures can be applied for routine quality control analysis of raloxifene in pharmaceutical dosage form.
Key words: raloxifene, analytical method, high performance liquid chromatography, validation.
INTRODUCCIÓN
Químicamente, el clorhidrato del raloxifeno ([6-hydroxy-2-(4-hydroxyphenyl)benzo[b] thien-3-yl]-[4-[2-(1-piperidinyl) ethoxy]phenyl]-, hydrochloride), es una molécula perteneciente a la familia de los benzotiofenos, la cual tiene la propiedad de ser un modulador selectivo del receptor estrogénico, por lo que se emplea en el tratamiento de la osteoporosis posmenopáusica. Una dosis de 60 mg de este medicamento al día incrementa la densidad mineral ósea total, similar al incremento que ocasionan los estrógenos conjugados con medroxiprogesterona o el alendronato en dosis de 5 mg/día. Sin embargo, a nivel de la densidad ósea de la espina lumbar los efectos del raloxifeno son inferiores a los de los otros dos fármacos.1-6
Diversos métodos de análisis para la determinación de raloxifeno puro y en formas farmacéuticas han sido informados en la literatura consultada.7-11 La cromatografía líquida de alta resolución, ha sido utilizada por ser un método simple, rápido, exacto y sensitivo, empleándose columna fase reversa y detección ultravioleta (UV) a longitud de onda de 286 ± 2 nm.8-10
En Cuba, se ha desarrollado un proceso tecnológico para la elaboración de tabletas de raloxifeno con el objetivo de introducir dicho medicamento como parte de la cartera de productos de la industria farmacéutica. Para garantizar el control de calidad de dichas producciones y los estudios de estabilidad correspondientes se estableció un método de análisis por cromatografía líquida de alta resolución.
El objetivo de este trabajo es validar el método de análisis desarrollado para comprobar que cumple con los parámetros establecidos en la literatura especializada12-14 y que sea factible su empleo como metodología de análisis en el control de la calidad que se realice a la formulación cubana del referido medicamento.
MÉTODOS
Se emplearon muestras correspondientes al lote 09002 de tabletas de raloxifeno de 60 mg, elaborado a escala piloto en la UCTB Tecnologías Básicas del Centro de Investigaciones y Desarrollo de Medicamentos (CIDEM).
MÉTODO DE ANÁLISIS
Para la determinación de raloxifeno en la muestra, se desarrolló un método de análisis por cromatografía líquida de alta resolución, basado en una modificación a la fase móvil realizada al método propuesto por Pavithra y Lakshmi.8
Se emplearon para ello las condiciones experimentales siguientes: precolumna Aluspher® 100 (RP-select B 5 mm) con columna Lichrospher® 100, RP 18 (25 cm de longitud y 4 mm de diámetro y 5 µ), utilizándose como fase móvil una mezcla de metanol-agua desionizada-trietanolamina (70:30:0,1) y detector UV a una longitud de onda de 286 nm. El flujo de la fase móvil fue de 1,5 mL/min y el volumen de inyección fue de 20 mL.
La preparación de las muestras se realizó de la manera siguiente: se pesó con exactitud la cantidad de muestra equivalente a 60 mg de ingrediente farmacéutico activo y se trasvasó a un matraz aforado de 100 mL de capacidad. Se adicionaron 40 mL de fase móvil y se agitó con ayuda de un baño ultrasónico por 10 min. Posteriormente se llevó a volumen con fase móvil, se mezcló y se filtró. Se tomaron con exactitud 2,0 mL de esta solución y se trasvasó a un matraz aforado de 25 mL de capacidad, llevándose a volumen con solución de fase móvil.
Paralelamente se preparó la solución estándar. Para ello se pesó con exactitud 15 mg de raloxifeno estándar y se trasvasó a un matraz aforado de 50 mL de capacidad. Se adicionaron alrededor de 30 mL de fase móvil y se agitó con ayuda de un baño ultrasónico por 10 min. Se llevó a volumen con fase móvil, se agitó y filtró. Se tomaron con exactitud 4,0 mL de esta solución, se trasvasó a un matraz aforado de 25 mL de capacidad y se llevó a volumen con fase móvil.
Todos los reactivos empleados fueron de calidad pura para análisis y la cristalería se encontraba apta para su uso.
VALIDACIÓN DEL MÉTODO DE ANÁLISIS
Se realizó un análisis de la especificidad del método. Para ello se colocaron muestras de materia prima, placebo y tabletas en las siguientes condiciones de trabajo:
- 70 ºC durante 72 h.
- Medio básico (NaOH 1 mol/L) y 70 ºC durante 72 h.
- Medio ácido (HCl 1 mol/L) y 70 ºC durante 72 h.
- Medio oxidante (peróxido de hidrógeno al 30 %) por 24 h.
Al finalizar se realizó el cromatograma por el método descrito, para comprobar si el pico de degradación se separó del pico de la muestra en las condiciones de trabajo establecidas.
En la determinación de la precisión del método se realizó la evaluación de la precisión intermedia; para lo cual se efectuaron 10 determinaciones a la concentración del 100 % por dos analistas diferentes. Con los resultados obtenidos se determinó la media, la desviación estándar y el coeficiente de variación para cada analista; además, se realizaron las pruebas de significación adecuadas para medias y varianzas (Fisher y Student) con el objetivo de determinar si existían diferencias significativas entre los resultados de cada uno de ellos.14
Para determinar la exactitud se trabajó con el método de análisis repetitivo de varias muestras de concentraciones diferentes conocidas. Para ello a partir de un placebo se prepararon cinco muestras por adición de cantidades exactas de analito patrón correspondiente al 80, 90, 100, 110 y 120 % del contenido teórico y cada muestra se analizó por triplicado acorde con el diseño experimental elaborado. Una vez realizada la experiencia, se determinó el porcentaje de recuperación para cada una de las concentraciones estudiadas.
Se determinó mediante la prueba t de Student si existían diferencias significativas entre el porcentaje de recuperación medio y el 100 % de recuperación; además, se calcularon si existía o no influencia del factor de concentración en los resultados, mediante la aplicación de la prueba de G de Cochran.14
Para la determinación de la linealidad se realizó la curva de calibración en la que se graficaron los valores de las respuestas (área) contra la concentración. Para ello se preparó una curva con concentraciones equivalentes a 42, 48, 54, 60, 66, 72 y 78 µg/mL de raloxifeno (equivalentes al rango comprendido entre el 70 y 130 %), realizándose la determinación según el método propuesto.
Se determinó mediante la prueba t de Student si el intercepto es o no significativo. Además, se aplicó la prueba de linealidad determinando el coeficiente de variabilidad de los factores de respuesta.
Para la determinación del intervalo se trabajó con concentraciones entre el 90 y el 110 % del contenido teórico a partir de las muestras preparadas para la determinación de la exactitud, y se determinó si en este intervalo se cumplían los criterios establecidos para la exactitud y la precisión.
En todos los casos se realizaron al menos tres repeticiones. Todos los análisis estadísticos se realizaron con el programa Statgraphics plus (versión 5.1, EUA). El nivel de significación establecido fue el de a £ 0,05.
RESULTADOS
Los resultados del estudio de especificidad del método demostraron que al realizarse el tratamiento en medio básico se observa en el cromatograma de las tabletas un pico de degradación a un tiempo retención de 9,0 min y una disminución en la intensidad del pico correspondiente al ingrediente farmacéutico activo. En el cromatograma se observa que en la muestra de ingrediente farmacéutico activo ocurrió lo mismo, mientras que en la muestra placebo no apareció ningún pico asociado a la posible degradación (Fig. 1).
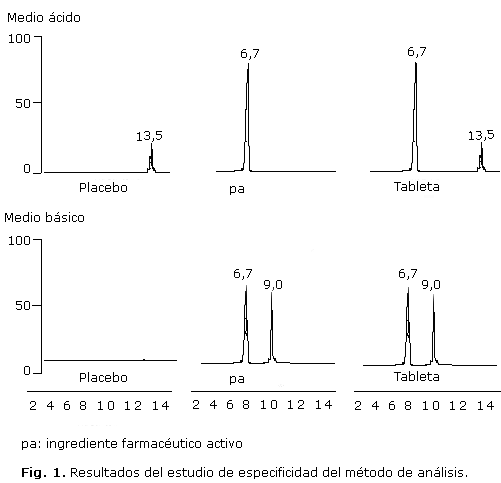
Por otro lado, al evaluar los resultados del tratamiento en medio ácido y 70 ºC durante 72 h, se observó la aparición de un pico de degradación a un tiempo de retención de 13,6 min en el cromatograma correspondiente a la muestra placebo tratada en igualdad de condiciones y no en el cromatograma correspondiente al ingrediente farmacéutico activo (Fig. 1), por lo que se puede atribuir a alguno de los excipientes presentes en la tableta y no a un producto de degradación del ingrediente farmacéutico activo. Se demuestró además, que es separado en las condiciones de trabajo propuesta.
Para el resto de los tratamientos propuestos no se observaron cambios en los cromatogramas obtenidos respecto al cromatograma del analito.
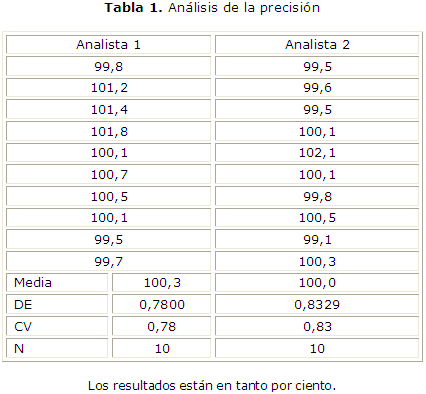
La precisión del método presentó los resultados que se muestran en la tabla 1, donde se observa que los coeficientes de variación están dentro de los límites establecidos para métodos cromatográficos (£ 2 %). Al aplicar la prueba de Fisher se obtuvo un valor de Fexp= 1,14 y una Ftab= 3,18 para F9/9 y p= 0,05, mientras que la prueba t de Student aplicada para comparar las medias de cada analista demostró la texp= 1,20 y la ttab= 2,10 para p= 0,05 y g.l.= 18.
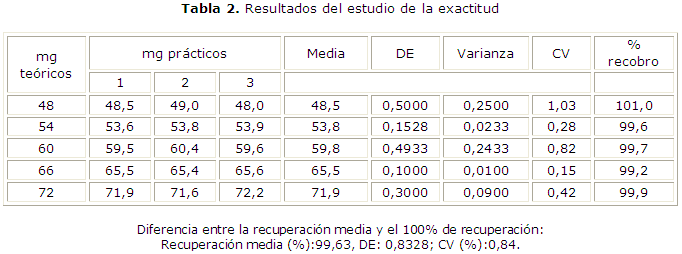
Los resultados de la exactitud se muestran en la tabla 2. La ecuación de la curva de recobro fue del tipo Y= 0,976X + 1,366 con un coeficiente de correlación igual a 0,999 (Fig. 2); al aplicar la prueba t de Student se obtuvieron los resultados de la t experimental del intercepto (texp= 1,31) y la t experimental de la pendiente (texp= 1,42) menor que la t tabulada de 2,14 para p= 0,05 y g.l.= 13.
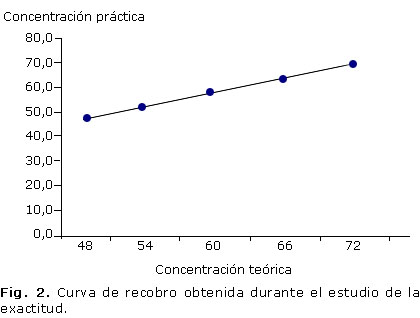
Al compararse mediante una prueba t de Student la recuperación media y 100 % de recuperación se obtuvo que la t experimental (texp= 1,72) es menor que la t tabulada (ttab= 2,09 para p= 0,05 y g.l.= 20). Por otro lado, al aplicarse la prueba de la G de Cochran se obtuvo un valor de la G experimental (Gexp= 0,4054) menor que la G tabulada (Gtab= 0,6838) para p= 0,05; K= 5 y n= 3.
En la evaluación de la linealidad del sistema se obtuvo una curva del tipo Y= 0,871X + 0,136, con un coeficiente de correlación de 0,999, que cumple con las exigencias para este tipo de análisis según se plantea en la literatura12,13 (Fig. 3). Al aplicar la prueba t de Student se obtuvo un valor de t experimental para el intercepto de 1,350; el valor de t tabulada resultó de 2,093 para n-2 grados de libertad y p= 0,05, mientras que la prueba de la proporcionalidad demostró que el intercepto es no significativo al obtenerse una texp= 0,0005, el cual es menor que la tteor= 2,093, para n-2 g.l. y p= 0,05 y no estadísticamente diferente de cero.
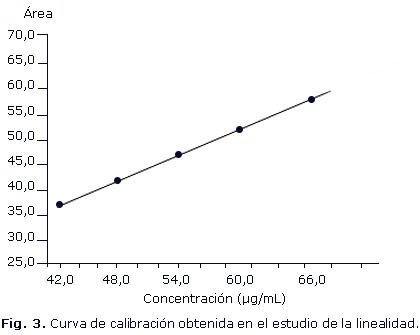
Por otro lado, el coeficiente de variación de los factores respuesta fue de 0,87 %, el cual es menor que el límite establecido de 5 %.
En el estudio del intervalo, al compararse mediante una prueba t de Student la recuperación media y 100 % de recuperación se obtuvo que la t experimental (texp= 1,90) es menor que la t tabulada (ttab= 2,31 para p= 0,05 y g.l.= 8). Por otro lado, al aplicarse la prueba de la G de Cochran se obtuvo un valor de la G experimental (Gexp= 0,859) menor que la G tabulada (Gtab= 0,871 para p= 0,05; K= 3 y n= 3).
DISCUSIÓN
Los resultados de los estudios de especificidad del método de análisis comprueban que en el medio básico el ingrediente farmacéutico activo sufre degradación, con la aparición de un pico a un tiempo de retención (9 min) diferente al pico del ingrediente farmacéutico activo, los cuales se separaron sin dificultad por el método de análisis propuesto. Estos resultados eran de esperar pues estudios realizados por Pérez y otros,11 encontraron que a valores de pH superiores a 7,5 el pico del cromatograma del raloxifeno disminuye, con la aparición de otro pico presumiblemente correspondiente al producto de degradación. Por otro lado, Srinivas y otros15 encontraron que al tratarse muestras de raloxifeno en medio básico (0,5 N NaOH), esta se degradaba con la consecuente formación de ácido 4[2-(piperidinel)etoxi]benzoico y N óxido de [2(4 hidroxifenil)-6-hidroxibenzo[b]tiel-3-il] [4-[2-(piperidinel)etoxi]fenil]metanona, denominadas en la literatura como impurezas D y A, respectivamente.15 La impureza mayoritaria es la D, la cual aparece en un tiempo de retención de aproximadamente 9 min.
Se comprueba además, que el medio ácido provoca la aparición de un pico de degradación, pero en este caso se corresponde con algunos de los excipientes presentes en la formulación, pues se conoce que en este medio el ingrediente activo no sufre degradación.15
Los resultados del tratamiento en medio oxidante no mostraron degradación del ingrediente activo en las condiciones de trabajo establecidas en este trabajo, aunque en los estudios de Srinivas y otros15 sí se informa que el medio oxidante provoca degradación con la consecuente formación de cloruro de 6 metilsulfoniloxi-2-[(4-metilsulfoniloxi)fenil]-3-[[4(2-(piperidinel)etoxi]benzol]benzotiofeno (denominada impurezas C) en forma de trazas, la cual aparece a un tiempo de retención de aproximadamente 8 min, muy alejado del tiempo de retención del ingrediente activo y fácilmente separable por el método de análisis propuesto.
Para el caso del tratamiento con calor (70 ºC) no se observaron cambios, aspecto que corrobora lo informado en la literatura,15 lo cual se demuestra que el ingrediente farmacéutico estudiado no se afecta por la temperatura. Por otro lado, no se realizaron los estudios de tratamiento con luz (254 nm), pues se ha informado que esta no afecta el raloxifeno.15
Por todo lo anterior, se puede afirmar que el método de análisis es específico para la determinación del ingrediente farmacéutico activo y sus productos de degradación, por lo que puede ser empleado para realizar el control de la calidad del medicamento.
Los resultados del análisis de la precisión demostraron que los valores del coeficiente de variación para cada analista están por debajo del 2 %, lo que es adecuado para métodos cromatográficos según la literatura.12-14
Al aplicar la prueba de Fisher para comparar las dispersiones se comprobó que no existen diferencias significativas entre estas. Iguales resultados se obtuvieron al compararse las medias mediante la prueba t de Student (tabla 1). Partiendo de estos resultados podemos considerar que las muestras son homogéneas, así se demostró así que el método desarrollado cumple con el parámetro de precisión.12-14
El análisis de los resultados del estudio de la exactitud mostró que en todos los casos los coeficientes de variación son menores al 2 % y los porcentajes de recuperación están entre 98 y 102 %, mientras que el valor experimental de la G de Cochran fue menor que el valor tabulado, lo que nos indica que las varianzas de las concentraciones empleadas son equivalentes; así se puede asumir que la concentración no influye en la variabilidad de los resultados.12-14
Los resultados de la diferencia entre la recuperación media y el 100 % de recuperación demostraron que no existían diferencias significativas entre ellas, al comprobarse que el valor de la texp es menor que el valor de la tteor.
La prueba de linealidad demostró que el coeficiente de variabilidad de los factores de respuesta es menor del 5 %, así se cumplimentó lo establecido para la significación de la pendiente y el intercepto, por lo que se puede afirmar que existe linealidad.
Se cumple además, que los coeficientes de variación en el estudio del intervalo y los porcentajes de recobros se encuentran dentro de las especificaciones establecidas, de manera que el valor de la G de Cochran resultó menor que el valor tabulado, por lo que también se cumple que las varianzas de las concentraciones empleadas son equivalentes, así se puede asumir que la concentración no influye en la variabilidad de los resultados. Este resultado es importante, pues se evaluó el intervalo que coincide con los límites de calidad establecidos para la concentración del principio activo en la forma terminada, lo que demuestra que dentro de ese intervalo el método propuesto cumple con los criterios de exactitud, precisión y linealidad.
Como conclusión se puede afirmar que el método desarrollado para la cuantificación por cromatografía líquida de alta resolución de raloxifeno en la formulación de tabletas cubana es específico, preciso, exacto y lineal, por lo que puede ser empleado para el control de la calidad del medicamento.
REFERENCIAS BIBLIOGRÁFICAS
1. Delmas PD, Bjarnason NH, Mitlak BH. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N Engl J Med. 1997;337:1641-7.
2. Ettinger B, Black DM, Mitlak BH, Knickerbocker RK, Nickelsen T, Genant HK, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA. 1999;282(7):637-45.
3. Lippman ME, Krueger KA, Eckert S, Sashegyi A, Walls EL, Jamal S, et al. Indicators of lifetime estrogen exposure: effect on breast cancer incidence and interaction with raloxifene therapy in the multiple outcomes of raloxifene evaluation study participants. J Clin Oncol. 2001;19(12):3111-6.
4. D'Amelio P, Muratore M, Tinelli F, Tamone C, Cosentino L, Quarta E, et al. Effect of raloxifene and clodronate on bone density in postmenopausal osteoporotic women. Int J Tissue React. 2003;25(2):73-8.
5. Weinstein RS, Parfitt AM, Marcus R, Greenwald M, Crans G, Muchmore DB. Effects of raloxifene, hormone replacement therapy, and placebo on bone turnover in postmenopausal women. Osteoporos Int. 2003;14(10):814-22.
6. Heringa M. Review on raloxifene: profile of a selective estrogen receptor modulator. Int J Clin Pharmacol Ther. 2003;41(8):331-45.
7. Basavarah K, Tharpa K, Rangachar U, Rajedraprasad N, Ganeshbahat S, Basavaiah K. Optimized and validated spectrophotometric methods for the determination of raloxifene in pharmaceuticals using permanganate. Arch Pharmacol Res. 2009;32(9):1271-9.
8. Pavithra DC, Lakshmi S. RP-HPLC estimation of raloxifene HCl in tablets. Indian J Pharm Sci. 2006;68:401-2.
9. Shabir GA. Validation of high-performance liquid chromatography methods for pharmaceutical analysis: Understanding the differences and similarities between validation requirements of the US Food and Drug Administration, the US Pharmacopeia and the International conference on Harmonization. J Chromatogr A. 2003;987:57-66.
10. Trontelj J, Tomaz V, Bogataj M, Mrhar A. HPLC analysis of raloxifene hydrochloride and its application to drug quality control studies. Pharm Res. 2005;52:334-9.
11. Pérez Ruiz T, Martinez Lozano C, Sanz A, Bravo E. Development and validation of a quantitative assay for raloxifene by capillary electrophoresis. J Pharm Biomed Anal. 2004;34(5):891-7.
12. United States Pharmacopoeia. USP 31. Washington DC.: United Pharmacopoeial Convention, Inc.; 2008. p. 752-7.
13. Chung Chow C. Analitycal method validation and instrument performance verification. Cap. 2. New Jersey: Ed. Wiley Interscience Sons; 2004. p. 11-26.
14. CECMED. Regulación No. 41. Validación de métodos analíticos. La Habana: CECMED; 2007.
15. Srinivas G, Kanumula G, Madhavan P, Kishore Kumar K, Rama Koti Reddy Y, Vishnu Priya M, et al. Development and validation of stability indicating method for the quantitative determination of Raloxifene hydrochloride and its related impurities using UPLC. J Chem Pharm Res. 2011;3(1):553-62.
Recibido: 16 de septiembre de 2013.
Aprobado: 30 de octubre de 2013.
Jorge E. Rodríguez Chanfrau. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). Ave. 26 No. 1605 entre Boyeros y Puentes Grandes. CP 10600. Plaza de la Revolución, La Habana, Cuba. Correo electrónico: jorge.rodriguez@infomed.sld.cu

{kind=link}