










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Farmacia
Print version ISSN 0034-7515
Rev Cubana Farm vol.48 no.2 Ciudad de la Habana Apr.-June 2014
PRODUCTO NATURAL
Validación del sistema ultramicroelisa en la certificación de placenta humana como materia prima farmacéutica y cosmética
Validation of the Ultra Micro Elisa for certification of human placent as a source of pharmaceuticals and cosmetics
MSc. Maydelin Trujillo Alfonso,I Lic. Sisley Rodríguez García,I MSc. Maria Teresa Pérez Guevara,II MSc. Kenia RomeroMartínez,II Téc. Yamilka Cala Mató,I Lic. Zoe Betancourt Valiente,I Téc. Reysel Santos MarreroI
I Planta Derivados de la Placenta. Laboratorio de Control Viral. La Habana, Cuba.
II Laboratorio de Investigaciones del SIDA. La Habana, Cuba.
RESUMEN
Introducción: el Laboratorio de Control Viral de la Planta Derivados de la Placenta realiza la certificación de la placenta humana como materia prima farmacéutica y cosmética mediante el sistema ultramicroanalítico.
Objetivo: validar el sistema ultramicroelisa de determinación de antígeno de superficie del virus de la hepatitis B, anticuerpos contra el virus de la hepatitis C y virus de inmunodeficiencia humana tipo 1 y 2 en muestras de suero de cordón umbilical.
Métodos: se realizó la calificación de la operación y la validación del desempeño analítico de los sistemas UMELISA HBsAg Plus, UMELISA HCV y UMELISA HIV 1+2 Recombinant empleandocomo sistemas de referencia el Hepanostika HBsAg Uni-FormII, Hepanostika HCV Ultra y Vironostika HIV-Uni-Form II Ag/Ab.
Resultados: la calificación de la operación para las tres técnicas analíticas resultó satisfactoria. Los parámetros de especificidad diagnóstica y analítica fueron de 100 %, así como la concordancia con las técnicas de referencia. El coeficiente de variación fue menor del 10 % durante el estudio de precisión interensayo, menor que el 20 % intraensayo y se demostró la robustez de las técnicas para pequeños cambios en la temperatura de incubación.
Conclusiones: los sistemas ultramicroelisa utilizados como método de control de la calidad de la placenta humana resultaron específicos, precisos y robustos en las condiciones ensayadas, por lo que pueden emplearse de manera segura y confiable.
Palabras clave: placenta humana, diagnosticadores, ELISA, validación, control de calidad.
ABSTRACT
Introduction: the Viral Control Laboratory of the Placenta Derivatives Production Plant certifies the human placenta as pharmaceutical and cosmetic raw material by means of the microultra analytic system.
Objective: to validate the Ultra Micro System for determination of hepatitis B surface antigen, antibodies to Hepatitis C Virus, and Immunodeficiency Human Virus Type 1 and 2 in umbilical cord serum samples.
Methods: the qualification of the operation and the validation of the analytic performance of the UMELISA HBsAg Plus system, UMELISA HCV and UMELISA HIV 1+2 Recombinant using the Hepanostika HBsAg Unite-Form II, Hepanostika Ultra HCV and Vironostika HIV-unite-Form II Ag/Ab as reference systems.
Results: the qualification of the operation for the three analytic techniques was satisfactory. The diagnostic and analytic specificities were 100 %; as well as the agreement with the reference techniques. The variation coefficient was lower than 10 % during the inter-assay precision study, below 20 % intra-assay and the robustness of the techniques was shown to manage small changes in the incubation temperature.
Conclusions: the ELISA Ultra Micro Systems used as quality control methods of the human placenta were specific, precise and robust under the tested conditions, so they can be safely and reliably used.
Key words: human placenta, diagnosticians, ELISA, validation, quality control.
INTRODUCCIÓN
En la Planta Derivados de la Placenta (DEPLACEN) se elaboran medicamentos y cosméticos a partir de la placenta humana.1,2 Teniendo en cuenta los requisitos establecidos para materias primas de origen biológico, la validación de las técnicas analíticas empleadas para su certificación es de vital importancia y constituyen una exigencia actual de las entidades regulatorias.3-5
El sistema ultramicroanalítico (SUMA) fue desarrollado por el Centro de Inmunoensayo (CIE) en la década del 80 y hasta la fecha ha quedado demostrado las múltiples ventajas del método.6 La tecnología incluye equipamiento, estuches de reactivos y software.7,8 Dentro de sus programas para el diagnóstico de enfermedades infecciosas el Laboratorio de Control Viral utiliza el ensayo inmunoenzimático cualitativo de determinación del antígeno de superficie del virus de la hepatitis B (UMELISA HBsAg Plus),anticuerpos contra el virus de la hepatitis C (UMELISAHCV de 3ra generación) y el virus inmunodeficiencia humana tipo 1 y 2 (UMELISAHIV 1+2 Recombinant).9
Los sistemas diagnóstico ultramicroelisa (UMELISA) para la detección de estos marcadores virales están validados por sus productores en muestras de suero o plasma humano y en sangre seca sobre papel de filtro.10-12 No existen evidencias de la validación de estos sistemas en muestras de suero de cordón umbilical, por lo que este es el objetivo del trabajo.
MÉTODOS
INMUNOENSAYOS
Para el pesquisaje de las muestras de suero de cordón umbilical se emplearon el UMELISA HBsAg Plus, UMELISA HCV, UMELISA HIV 1+2 Recombinant y como sistemas de referencia el Hepanostika HBsAg Uni-FormII, Hepanostika HCV Ultra y el Vironostika HIV-Uni-Form II Ag/Ab. Para el estudio de los resultados discordantes se emplearon los sistemas confirmatorios Hepanostika HBsAg UniForm II Confirmatory, Inno LIA HCV Score y DAVIH-BLOT.
MUESTRAS
Para la validación de los sistemas se emplearon muestras obtenidas por centrifugación a partir de la sangre de cordón umbilical, recogidas durante el alumbramiento en los salones de parto de los hospitales ginecobstétricos incluidos en el Sistema Nacional de Recogida de Placenta, según lo establecido en la Regulación No. 2 del 2002 del Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED) "Placenta humana como materia primafarmacéutica".13
Las muestras positivas a los marcadores virales del antígeno de superficie del virus de la hepatitis B (HBsAg), anticuerpos contra el virus de la hepatitis C(anti-HCV) y contra el virus inmunodeficiencia humana tipo 1 y 2 (anti-HIV)fueron preparadas en el LISIDA a partir de un pool de muestras de suero de cordón umbilical negativas y caracterizadas por los sistemas de referencia. Se obtuvieron diferentes niveles de reactividad desde bajos, medios y fuertes.
CALIFICACIÓN DE LA OPERACIÓN
Se realizó mediante la comprobación de la existencia de procedimientos normalizados de operación (PNO) adecuados e idóneos para la ejecución de las técnicas analíticas UMELISA y la preparación de soluciones. La calificación del personal se verificó mediante la revisión del Expediente Técnico comprobando la existencia de Curso de acreditación para el empleo de la tecnología SUMA y/o adiestramiento inicial y específico en el puesto de trabajo. Los equipos e instrumentos necesarios para la realización de las técnicas (incubadora BINDER, multipipeta Erizo Programable 202, lavadores MW-201, lector de placas UMELISA PR- 521, micropipetas y cristalería) se verificaron mediante la presencia de etiqueta de apto para el uso y la vigencia de la fecha de calibración y/o verificación. Se revisó el cumplimiento de los planes de mantenimiento y calibración.14
CALIFICACIÓN DEL DESEMPEÑO
Se realizó la validación de las técnicas analíticas a través de los protocolos de validación para los tres sistemas UMELISA evaluando especificidad, precisión, robustez y el límite de detección.15-17
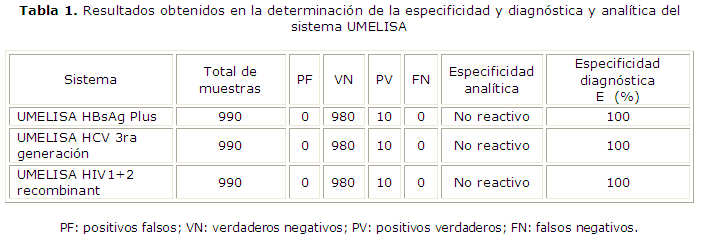
Especificidad: se realizó un diseño experimental de 980 muestras negativas y 20 positivas con factores de interferencia cruzada (anti-HIV, anti-HCV y HBsAg), alternandolos en dependencia de la técnica a validar. Se evaluó especificidad diagnóstica (E) con un criterio de E≥ 99 % y la especificidadanalítica.18
Estudio de concordancia: se compararon los resultados de las 980 muestras negativas y de 10 muestras positivas con diferentes niveles de reactividad. Se calculó la concordancia de los resultados obtenidos entre los sistemas de referencia y los sistemas UMELISA utilizando el índice Kappa con un criterio de aceptación entre 0,61 y 1 que son los criterios de "bueno" y "muy bueno" respectivamente y el porcentaje.18
Para el análisis de los resultados se confeccionaron tablas de contingencia de doble entrada.
Precisión: se evaluó a través de la repetibilidad con un diseño experimental diferenciado por técnica. Para obtener muestras con niveles de reactividad media para los tres marcadores virales, se prepararon las siguientes diluciones: UMELISA HBsAg: dilución de 1/1 000 a partir de una muestra con título 2154 UI/mL, UMELISA HCV: dilución 1/20 a partir de una muestra con elevado nivel de reactividad, UMELISA HIV 1+2 Recombinant: dilución 1/60 a partir de una muestra con alto título de anticuerpos. Se ensayaron 30 réplicas, por un mismo analista, con el mismo lote de reactivos, el mismo día.
Para la precisión intermediase analizaron cuatro réplicas de una muestra con reactividad alta, media y baja para los tres sistemas UMELISA en tres días, utilizando tres lotes de reactivos diferentes.
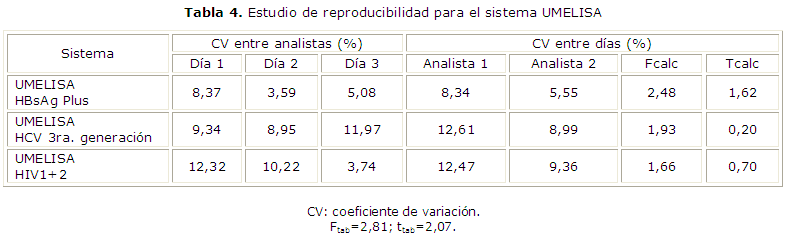
En el estudio de reproducibilidad se estudiaron cuatro réplicas de una muestra con reactividad media por un analista en el Laboratorio DEPLACEN y en el LISIDA en tres días diferentes con el mismo lote de reactivos. Se aplicaron las pruebas de Fisher y de la t de Student para determinar si existían diferencias significativas entre los resultados al variar las condiciones de análisis.
Se calculó la media, desviación estándar (DE) y coeficiente de variación (CV) entre réplicas de la misma muestra para los ensayos de repetibilidad, para los diferentes días y lotes en la precisión intermedia y entre días y analistas para la reproducibilidad. Los datos fueron analizados mediante el programa Excel de Microsoft Office.
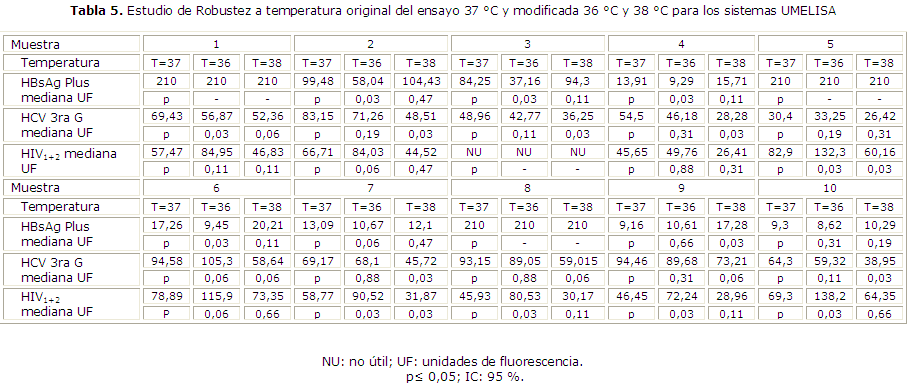
Robustez: se analizaron 10 muestras positivas para cada marcador viral. Se ensayaron cuatro réplicas de cada muestra; se realizó el estudio utilizando como control la temperatura recomendada por el fabricante (37 °C) y las variaciones 36 y 38 °C en la etapa de incubación de las muestras y el conjugado. Se colocaron en la misma placa muestras reactivas y no reactivas en forma alterna, teniendo en cuenta las posibles contaminaciones cruzadas.
Se compararon las medianas de dos muestras (ensayo original y ensayo modificado) empleando la prueba de Mann-Whitney mediante el programa Statgraphics Plus (SGWIN. Copyright© 2000). Se tomó como criterio un valor de p≤ 0,05 de significación estadística con un nivel de confianza de 95 %.
Límite de detección: se realizó un diseño experimental diferenciado por técnica empleando cuatro réplicas de las diluciones: UMELISA HBsAg: dilución de 1/500 hasta 1/20 000 a partir de una muestra con título 2154 UI/ mL, UMELISA HCV y UMELISA HIV1+2: dilución 1/2 hasta 1/4096 a partir del estándar de referenciacon un nivel de reactividad media. Se estableció como límite de detección la última dilución en la que se detectó la presencia del marcador viral.18
RESULTADOS
CALIFICACIÓN DE LA OPERACIÓN
La calificación de la operación para las tres técnicas analíticas resultó satisfactoria ya que los procedimientos relacionados con la ejecución de las técnicas y preparación de soluciones fueron adecuados, el personal estaba calificado y entrenado para realizar las operaciones y los equipos se encontraban calibrados y aptos para el uso.
CALIFICACIÓN DEL DESEMPEÑO
La especificidad diagnóstica fue de 100 % para los sistemas UMELISA HBsAg Plus, UMELISA HCV y UMELISA HIV 1+2 Recombinant al comparar los resultados de las 980muestras negativas con los sistemas de referencia Hepanostika HBsAg Uni-FormII, Hepanostika HCV Ultra, Vironostika HIV-Uni-Form II Ag/Ab. Los tres sistemas analizados fueron específicos en la identificación cualitativa de su marcador viral exclusivamente (tabla 1).
Se obtuvo un 100 % de concordancia y un índice kappa de1 en los sistemas UMELISAHBsAg Plus, UMELISA HCV y UMELISA HIV 1+2 Recombinant, lo que indica una correlación "muy buena" entre los resultados observados y los esperados para el sistema UMELISA cubano y el sistema de referencia utilizado (tabla 2).

Los resultados del estudio de precisión se presentan las tablas 3 y 4. En el estudio de repetibilidad los CV obtenidos fueron menor que el 10 % mientras que en las dos variantes estudiadas de precisión intermedia y reproducibilidad fueron menor que el 20 %, catalogados como adecuados para este tipo de inmunoensayos. Los valores de F y t calculadas resultaron menores que los valores tabulados para cada uno de los niveles estudiados; los valores calculados para la prueba de Fischer fueron 2,48; 1,93 y 1,66; y en la prueba t de Student 1,62; 0,20 y 0,70 para los sistemas UMELISA HBsAg, HCV y HIV 1+2 respectivamente para un 95 % de confianza.
En el estudio de la robustez del sistema UMELISA HBsAg Plus y UMELISA HIV 1+2 Recombinant no hubo diferencias significativas en la reactividad de las 10 muestras positivas a 38 °C, de esta manera quedó demostrada la capacidad de los ensayos para brindar resultados consistentes a esta temperatura, no resultó así cuando la temperatura varía a 36 °C. Para el sistema UMELISA HCV no existieron diferencias significativas en la reactividad de las muestras positiva sala temperatura de 36 °C y sí hubo diferencias significativas a 38 °C para un nivel de confianza de 95 % (tabla 5).
El límite de detección en nuestras condiciones de trabajo fue de 1/10 000 para el sistema UMELISA HBsAg Plus, 1/64 para el sistema UMELISA HCV y 1/128 para el sistema UMELISA HIV 1+2 Recombinant.
DISCUSIÓN
La calificación de la operación se demostró mediante evidencia documental de la acreditación por el CIE del personal que desarrolla las técnicas analíticas, así como de la superación en temas relacionados (inmunoquímica, inmunología celular, buenas prácticas de laboratorio). El adiestramiento inicial y continuo en el puesto de trabajo con sus respectivas evaluaciones de "satisfactorias" sirvieron de argumento para demostrar que el personal está calificado y entrenado para realizar las operaciones. Se constató el cumplimiento de plan de calibración realizado en el centro y el plan de mantenimiento preventivo por el CIE. Todo el equipamiento incluyendo instrumentos de medición estaba calibrado, verificado y apto para el uso.
Los resultados obtenidos en la calificación del desempeño en cuanto a especificidad diagnóstica concuerdan con lo reportado por los productores para el estuche UMELISA HIV 1+2 en muestras de suero de donantes de sangre, pacientes infestados con ambos tipos de virus, sin relación con este y en muestras de sangre seca sobre papel de filtro: pacientes seropositivos y donantes de sangre.12 En cuanto al UMELISA HCV hay coincidencia con los estudios realizados por el CIE en muestras de suero de plasmaféresis y en muestras de sangre seca sobre papel de filtro11 y con otra investigación realizada en pacientes multitransfundidos, pero en este caso utilizando como referencia el UVI® HCV EIA 4.0.19 La E obtenida en este estudio fue superior a la alcanzada con muestras de donantes de sangre y hemodializados que informan 99,93 % y 99,33 % respectivamente.11 Para el sistema UMELISA HBsAg Plus los resultados fueron similares a los obtenidos por los productores en muestras de sangre seca sobre papel de filtro y mayor para muestras de suero o plasma en que indican una especificidad del método de 99,92 %.10 Otro estudio de evaluación de desempeño del InmunoLISATM,20 utilizando como referencia el propio UMELISA HBsAg Plus informa valores de E iguales a los nuestros.
La variación de un diagnosticador respecto a un estándar (o prueba de oro), se mide por la concordancia alcanzada al examinar y clasificar una serie de muestras biológicas. Para los tres sistemas UMELISA, comparados con los sistemas de referencia, se obtuvo una muy buena correlación con un valor de K= 1, índice este superior al informado en un estudio con el UMELISA HCV 3ra Gen en muestras de pacientes multitransfundidos (k= 0,9582)19 y otra investigación de desempeño del DAVITH-VIH 2 en que se obtuvo un valor de K= 0,978.21 La prueba ELISA ha mostrado una concordancia hasta 98,3 %, tanto en sueros positivos como negativos en estudios comparativos entre diferentes técnicas para la determinación del HBsAg, la cual es significativamente mayor que la observada en otras pruebas como el ensayo inmunoenzimático de micropartículas (90 %) y la hemaglutinación (89 %).22
Es conocida la falsa reactividad de los ensayos inmunoenzimáticos en individuos con situaciones no relacionadas con el marcador viral estudiado, los cuales son propensos a causar reacciones cruzadas. En la detección de anticuerpos al VHC esta puede deberse entre otras causas a infecciones virales con el HIV y el virus de la hepatitis B,23 lo que ha sido corroborado por otros autores.24 En este estudio no se obtuvieron resultados reactivos en las muestras que pudieran provocar reacciones cruzadas lo que demuestra la especificidad analítica de los tres sistemas UMELISA y su capacidad como métodos de control de calidad de la placenta en el Laboratorio de Control Viral.
En el estudio de repetibilidad para el sistema UMELISA HBsAg Plus se obtuvo un coeficiente de variación óptimo y para el sistema UMELISA HCV y UMELISA HIV 1+2 se alcanzó un coeficiente de variación adecuado, lo que demuestra la buena precisión de los tres sistemas analizados. En el estudio de precisión intermedia y reproducibilidad los CV estuvieron entre el 3,59 a 17,59 % (HBsAg Plus), 7,87 a 19 % (HCV) y 3,26 a 18,87 % (HIV 1+2). La variabilidad de los resultados se encuentra dentro de los límites según lo establecido por el CECMED,3,5 quien plantea que en los inmunoensayos enzimáticos el coeficiente de variación no debe superar el 10 % en la prueba de precisión intraensayo, el 20 % en la interensayo y se consideran óptimos los inferiores al 5 % y 10 % respectivamente. Similares resultados han sido publicados por otros autores.23,24
El análisis de la reproducibilidad por las pruebas de Fischer y t de Student, demuestra que no existen diferencias significativas entre las precisiones alcanzadas por los analistas en diferentes días para un 95 % de confianza, ya que el valor de F calculado fue menor que la F tabulada; estos resultados permiten establecer que las precisiones son similares. Al realizar las pruebas t de Student, el valor calculado resultó menor que el tabulado, para un 95 % de confianza, lo cual demuestra que no existen diferencias significativas entre las medias alcanzadas, con un nivel de significación del 5 %.
En relación a la robustez de los sistemas de ensayo quedó demostrado que la temperatura influye directamente en la consistencia del resultado analítico. Aunque los fabricantes informan 37 ± 1 °C para los tres sistemas UMELISA,10-12 en nuestras condiciones de trabajo el sistema UMELISA HBsAg Plus y HIV 1+2 fue robusto a 38 °C y el sistema UMELISA HCV 3ra Gen a 36 °C. Este resultado puede explicarse por el efecto de la temperatura en las reacciones enzimáticas; una elevación de esta incrementa la energía cinética de las moléculas reactantes, lo que provoca un aumento de colisiones productivas por unidad de tiempo hasta un punto (temperatura óptima de reacción), por encima del cual el incremento de la velocidad de la reacción puede ser anulado debido a la pérdida de la estructura tridimensional de la enzima25 por ruptura de los enlaces débiles de hidrógeno que sostienen su estructura secundaria y terciaria.
Respecto al límite de detección del HBsAg, los resultados se encuentran dentro de los requerimientos de la FDA(0,2-1 UI/mL) para este tipo de ensayos diagnóstico.20
Los sistemas UMELISA HBsAg Plus, UMELISA HCV y UMELISA HIV 1 + 2, demostraron su validez como métodos cualitativos de identidad, por lo que pueden emplearse de manera confiable y segura en muestras de suero de cordón umbilical.
REFERENCIAS BIBLIOGRAFICAS
1. Miyares C. Certificado de Inscripción Melagenina Plus. Registro Sanitario No 0985. Centro Estatal para el Control de Medicamentos (CECMED). Ministerio de Salud Pública. La Habana, Cuba. 1998.
2. Miyares C. Jabón bioactivante dérmico con ácido hialurónico. Registro Sanitario No 060/02,2002. Instituto de Nutrición e Higiene de los Alimentos. Ministerio de Salud Pública. La Habana, Cuba. 2002.
3. Regulación 41/07. Centro Estatal para el Control de Medicamentos (CECMED). Validación de métodos analíticos.Ministerio de Salud Pública. La Habana, Cuba.2007. [citado 13 Nov 2012]. Disponible en: http://www.cecmed.sld.cu/Pages/Reg_EvalEL.html
4. Regulación 37/12. Buenas Prácticas de laboratorio para el Control de Medicamentos (CECMED). Ministerio de Salud Pública. La Habana, Cuba. 2012. [citado 13 Nov 2012]. Disponible en: http://www.cecmed.sld.cu/Pages/Reg_EvalEL.html
5. Regulación 47/2007. Requisitos para la evaluación del desempeño de diagnosticadores. Centro Estatal para el Control de Medicamentos (CECMED). Ministerio de Salud Pública. La Habana, Cuba. 2007. [citado 13 Nov 2012]. Disponible en http://www.cecmed.sld.cu/Docs/RegFarm/DRA/Diag/Reg/Reg_47-07.pdf
6. Tecnosuma Internacional SA©. [Homepage on the Internet]. Sistema Ultra Micro Analítico (SUMA). [citado 13 Nov 2012]. Disponible en: http://www.tecnosuma.com
7. García MA, Carlos N. Producen nueva generación de lectores SUMA. Innovación, Ciencia y Desarrollo. 1997;3(3):41.
8. Díaz W, Carlos N, Rego A, Fernández JL. Software para trabajo con estuches UMELISA y lectores PR de la tecnología SUMA (Strips Reader Software SRS Ver 8.0). Registro con número I 0010132021200. Centro de Control Estatal de Equipos Médicos (CCEEM). La Habana, Cuba. 2001.
9. Vigilancia Epidemiológica. 2012. [citado 13 Nov 2012]. Disponible en: http://www.tecnosuma.com/vigilancia.html
10. Inserto del producto UMELISA HBsAg Plus. Centro de Inmunoensayo (CIE). La Habana, Cuba. 2006.
11. Inserto del producto UMELISA HCV. Centro de Inmunoensayo (CIE). La Habana, Cuba. 2004.
12. Inserto del producto UMELISA HIV1+2. Centro de Inmunoensayo (CIE). La Habana, Cuba. 2007.
13. Regulación 2/02. Placenta humana como materia prima farmacéutica. Centro Estatal para el Control de Medicamentos (CECMED). Ministerio de Salud Pública. La Habana, Cuba. 2002.
14. PVTAV-08-41. Protocolo de validación de las técnicas del laboratorio de Control Viral. 2008.
15. PVTAV-08-42. Protocolo de validación del método analítico UMELISA para la determinación de marcadores virales al HBsAg en muestras de suero de cordón umbilical. 2008.
16. PVTAV-08-43. Protocolo de validación del método analítico UMELISA para la determinación de marcadores virales al VHC en muestras de suero de cordón umbilical. 2008.
17. PVTAV-08-44. Protocolo de validación del método analítico UMELISA para la determinación de marcadores virales al VIH 1+2 en muestras de suero de cordón umbilical. 2008.
18. Ochoa R, Martínez J, Ferriol X, García A, Estrada E, Blanco R, et al. Guía para la estandarización de técnicas inmunoenzimáticas en ensayos de vacunas. VacciMonitor. 2000;9(3):13-8.
19. Rivero Jiménez RA, Merlín Linares JC, Blanco de Armas M, Navea Leyva LM, Lam Díaz RM, Castillo González D, et al. Eficacia diagnóstica de sistemas de inmunoensayos para el virus de la hepatitis C en muestras de pacientes multitransfundidos. Rev Cubana Hematol Inmunol Hemoter. 2009; 25(3):56-65.
20. Basualdo-Sigales MC, Alcántara-Pérez PA, Soler-Claudin C. Evaluación del estuche de diagnóstico InmunoLISATM HBsAg 1 step fabricado por Orgenics LTD para la detección del antígeno de superficie del virus de la hepatitis B. Bioquimia. 2008;33(3):122-7.
21. Martín Alfonso D, Silva Cabrera E, Pérez Guevara MT, Díaz Herrera DF, Romero Martínez K, Díaz Torres HM, et al. Diseño y evaluación del sistema DAVIH VIH-2. Rev Cubana Med Trop. 2007;59(3):241-6.
22. Beltrán M, Ayala M. Evaluación externa de los resultados serológicos en los bancos de sangre de Colombia. Pan Am J Public Health. 2003;13(2/3):138-43.
23. Colin C, Lanoir D, Touzet S, Meyaud-Kraemer L, Bailly F, Trepo C. Sensitivity and specificity of third-generation hepatitis C virus antibody detection assays: an analysis of the literature. J Viral Hepatitis. 2001;8(2):87-95.
24. Blanco-Rivero MC, Medrano G, García M, Capriotti G, Torruela M. Evaluación de un nuevo inmunoensayo para detectar anticuerpos contra el virus de la hepatitis C. Acta Bioquím Clin Latinoam. 2008;42(3):325-32.
25. Chávez MA, Díaz J, Pérez U, Delfín J. Efectos del pH y la temperatura. En: Chávez MA, Díaz J, Pérez U, Delfín J. Temas de enzimología. La Habana: Universidad de La Habana, Facultad de Biología; 1990. p. 287-90.
Recibido: 19 de diciembre de 2013.
Aprobado: 20 de enero de 2014.
Maydelin Trujillo Alfonso. Planta Derivados de la Placenta. Laboratorio de Control Viral. Autopista Novia del Mediodía y 173, Valle Grande, La Lisa. La Habana, Cuba. Correo electrónico: mayde@infomed.sld.cu

{kind=link}
{kind=link}
{kind=link}
{kind=link}