










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La enfermedad pulmonar intersticial (EPI) abarca un grupo de afecciones con manifestaciones clínicas, radiológicas y funcionales respiratorias similares; sus causas son muy variadas y en la actualidad se conocen más de 150 orígenes diferentes. De las neumonías intersticiales idiopáticas, se distinguen otras dos clases de EPI: de causa conocida o asociadas con otras entidades clínicas definidas, y las primarias o asociadas a otras enfermedades no bien definidas.1
La radiografía de tórax es un método insustituible en la evaluación inicial y el seguimiento de los pacientes con EPI. Los datos radiológicos relacionados con las EPI son el vidrio deslustrado, los patrones nodulillar reticular y reticulonodular, y el pulmón en panal de abejas. La prueba funcional respiratoria (PFR) es un elemento básico para cuantificar la gravedad y conocer la evolución de la enfermedad y la respuesta al tratamiento. El análisis celular e inmuno-histoquímico del lavado broncoalveolar (LBA) es de gran interés en la valoración diagnóstica de la EPI, sin embargo, se explora solo una región del pulmón y es poco probable que los hallazgos que puedan encontrarse traduzcan los cambios inflamatorios que afectan de forma difusa al parénquima pulmonar. En las enfermedades del tejido conectivo (ETC), los fenómenos de autoinmunidad originan una lesión de la célula del alveolo pulmonar que desencadena una cascada de fenómenos intermedios complejos que llevan a un depósito de material colágeno que provoca fibrosis pulmonar. Ciertos tratamientos utilizados en las enfermedades reumatológicas también pueden provocar lesiones fibróticas pulmonares.2
Las ETC que con mayor frecuencia presentan EPI a lo largo de su evolución incluyen al lupus eritematoso sistémico, artritis reumatoide, esclerosis sistémica, polimiositis/dermatomiositis, síndrome de Sjögren, enfermedad mixta del tejido conectivo y la espondiloartritis anquilosante.3,4
Se pueden diferenciar dos tipos de EPI; en la primera predomina la inflamación y puede tratarse con fármacos inmunomoduladores, en la segunda predomina la fibrosis pulmonar en la que estos fármacos no tienen ninguna utilidad. La forma fibrosante se comporta parecida a la fibrosis pulmonar idiopática, es también una afectación crónica, progresiva y mortal.5
La esclerosis sistémica es una enfermedad autoinmune del tejido conjuntivo, que se caracteriza por una desregulación inmunitaria y una fibrosis progresiva que afecta típicamente a la piel, con afectación variable de los órganos internos. Es una afección rara que afecta principalmente a mujeres jóvenes y de mediana edad, lo que resulta en una morbilidad y mortalidad desproporcionada. Actualmente, la enfermedad pulmonar intersticial es la causa de muerte más común entre los pacientes con esclerosis sistémica, con una prevalencia de hasta el 30 % y una mortalidad a 10 años de hasta el 40 %.6
El síntoma más común es la disnea con el ejercicio, otros son: tos, fatiga y dolor torácico. Al examen físico se suelen encontrar a nivel de las bases crepitantes en velcro; no obstante, un gran porcentaje de pacientes permanecen asintomáticos y el compromiso pulmonar ya está avanzado cuando se evidencian los hallazgos en el examen físico, lo que afecta negativamente la morbimortalidad.7
En la actualidad el diagnóstico se realiza mediante tomografía pulmonar computarizada de alta resolución (TACAR). El patrón observado más frecuente es la combinación de opacidades en vidrio deslustrado y reticulación fina, características de la neumonía intersticial no específica (NINE). Se han propuesto esquemas de estratificación simplificados basados en la definición de enfermedad extensa, cuando la extensión de la fibrosis supera el 20 % en el TACAR o si la capacidad vital forzada (CVF) es inferior al 70 %. La TACAR permite la detección de la neumopatía, incluso en estadios muy iniciales en individuos asintomáticos y que tienen una radiografía de tórax normal. Además, resulta útil para valorar la extensión de las lesiones parenquimatosas y su naturaleza.8
El hallazgo característico en la espirometría es un patrón restrictivo, con disminución de la capacidad vital forzada (CVF), pero son normales el volumen espiratorio en el primer segundo (VEF1) y la relación VEF1/CVF; la difusión de monóxido de carbono (DLCO), por su parte, se encuentra disminuida, dado que hay un engrosamiento del intersticio que disminuye el intercambio; esto suele ocurrir muy tempranamente.9
Se ha visto que el engrosamiento pleural es muy característico en la enfermedad pulmonar intersticial difusa (EPID) por ES, por lo que la ecografía ha surgido como alternativa para el diagnóstico temprano, con la ventaja de disminuir la exposición a la radiación; en los diferentes estudios se ha encontrado que un engrosamiento pleural mayor de 2,8-3 mm en varias zonas pulmonares es muy sugestivo de ES como etiología de la EPID. Otros hallazgos como las líneas B en el parénquima pulmonar, aunque presentes en un alto porcentaje de pacientes con compromiso intersticial por ES, no son específicos de la enfermedad.10,11
Métodos
Se realizó un estudio observacional, descriptivo y de corte transversal durante el período comprendido entre diciembre de 2018 y diciembre de 2019, en el servicio de reumatología del Hospital Clínico Quirúrgico Docente “Hermanos Ameijeiras” para caracterizar la enfermedad pulmonar intersticial en pacientes con esclerosis sistémica.
El universo del estudio se constituyó por 168 pacientes, los cuales se encuentran registrados en la consulta protocolizada de la institución en el período de estudio.
La muestra se obtuvo con la aplicación de los criterios de inclusión y exclusión.
Se incluyeron los pacientes con edad igual o mayores de 19 años, cumplir con los criterios de esclerosis sistémica del Colegio Americano de reumatología (ACR, por sus siglas en inglés) de 1980 (anexo) y pacientes con compromiso pulmonar por enfermedad pulmonar intersticial.
Se excluyeron los pacientes con historias clínicas con datos incompletos y los pacientes que no dieron su consentimiento para participar en el estudio. La muestra quedó conformada por 55 pacientes que cumplieron los criterios de inclusión establecidos, se emplearon muestreo no probabilístico de carácter intencional o a conveniencia.
La información se obtuvo a partir de los informes de resultados de tomografía de tórax y pruebas de función respiratoria, con las variables que dieron respuesta a los objetivos, además, toda la información fue recolectada del expediente clínico individual de cada paciente. Se confeccionaron tablas de distribuciones de frecuencia y se calculó el porcentaje en todas las variables. Se emplearon medidas de tendencia central como valor promedio para las edades y medida de dispersión absoluta como la desviación típica; la prueba ji cuadrado para verificar asociación significativa entre las variables involucradas, se utilizó el nivel de significación del 5 % (p ˂ 0,05).
Resultados
Se muestra la distribución de pacientes según edad, sexo y color de la piel. En cuanto a la edad, fue más frecuente en pacientes ˃ de 40 años, que representaron el 89,1 % del total, la edad promedio de los pacientes en estudio fue de 45,01 ± 11,8 años, con predominio el sexo femenino 96,4 % y piel mestiza para un 63,6 % (tabla 1).
Tabla 1 -. Distribución de pacientes según variables sociodemográficas
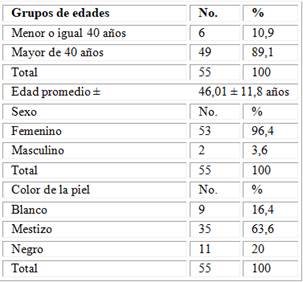
Fuente: Base de datos del Servicio de Reumatología.
La distribución de los pacientes según formas clínicas de la esclerosis sistémica. Se observa un predominio de la forma difusa en el 90,9 % de los casos sobre 9,1 % de la forma limitada (tabla 2).
Tabla 2 - Distribución de pacientes según formas clínicas
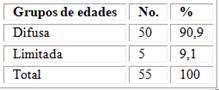
Fuente: Base de datos del Servicio de Reumatología.
Se muestra la distribución de pacientes según los patrones tomográficos en que se presenta la enfermedad pulmonar intersticial, se observó un predominio del patrón en panal en el 41,8 de los casos, seguido del patrón en vidrio esmerilado para un 36,4 % (tabla 3).
Tabla 3 - Patrones radiológicos por tomografía encontrados en este estudio
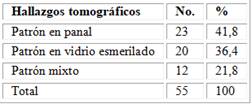
Fuente: Base de datos del Servicio de Reumatología.
Se muestra la distribución de los pacientes con ES y EPI según la CVF; se observó que más del 80 % (83,6 %) de estos, tenían una capacidad vital forzada disminuida (tabla 4).
Tabla 4 - Distribución de los pacientes con ES y EPI según la CVF
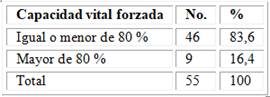
Fuente : Base de datos del Servicio de Reumatología.
La relación entre la capacidad vital forzada y anti-Scl-70. Los pacientes con anti-Scl-70, presentaron una mayor pérdida de la CVF al final del seguimiento, representaron un 40,0 % del total analizado, esto traduce una mayor proporción de patrón restrictivo en los pacientes con estos autoanticuerpos. El valor de p = 0,041 obtenido, garantiza la significación estadística de las relaciones existentes entre estas variables (tabla 5).
Tabla 5 - Relación entre la capacidad vital forzada y anti-Scl-70
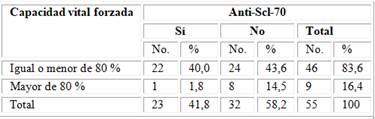
Fuente: Base de datos del Servicio de Reumatología. p = 0,041.
La relación entre la CVF y hallazgos tomográficos. Se muestra la distribución de pacientes según capacidad vital forzada y los hallazgos obtenidos en el TACAR, se observó la diversidad de los resultados en este procedimiento, lo que se evidencia en un valor de p = 0,997 que nos muestra una relación no significativa entre estas variables (tabla 6).

Discusión
Se han identificado diferentes factores asociados al deterioro de la funcion pulmonar, de ahí la importancia de reconocer e identificar a los enfermos con presencia de éstos, para poder intervenir, con el objetivo de frenar la progresión a la fibrosis y proporcionar al paciente un mejor estado de salud.
En el estudio la mayoría de los pacientes tenían más de 40 años. Estos resultados son similares al estudio de Arbeláez Solera y otros12 donde la edad promedio fue 46 años, con una mediana de 47 años ± 12 años. Al igual que lo reportado en Europa y Estados Unidos donde la edad promedio es de 47,9 y 46 años respectivamente.13,14 La distribución de los pacientes según sexo, con ES que presentaron EPI, fue más frecuente en el sexo femenino, la proporción mujer-hombre en el estudio fue elevada, es difusa para 23 mujeres por cada 1 hombre. En Arbeláez Solera y otros12 se señala que, en cuanto a la distribución por sexo, las mujeres son las más afectadas. Se encontró una relación por sexo y compromiso intersticial pulmonar mujer: hombre de 9:1.
Son pocos los estudios donde se determina la asociación entre el color de la piel y la EPI. En relación con lo propuesto por Silver y otros,15 plantean que la EPI se ha relacionado con pacientes afrodescendientes, cuando coexiste con la ES suele afectar más a las mujeres, y cuando se presenta en hombres es más grave, lo que podría sugerir que el paciente afroamericano tiene un ambiente de citocinas profibróticos que lo predispone a desarrollar EPI y un peor pronóstico.16
Los resultados en este estudio son similares a los de la investigación de Arbeláez Solera y otros12 en el cual, el compromiso pulmonar intersticial es más frecuente en pacientes con esclerosis sistémica difusa. Solo el 60 % de los pacientes presentan síntomas respiratorios a lo largo del curso de la enfermedad. Muchos se encuentran asintomáticos desde el punto de vista respiratorio, lo que motiva a realizar un interrogatorio dirigido, se buscan signos y síntomas para detectar de manera temprana la afectación pulmonar.
En el estudio de Hant y otros,7 la disnea, resulta ser el síntoma principal de compromiso pulmonar, similar a lo reportado por Walker y otros,17 que evaluaron el compromiso pulmonar intersticial en 60 pacientes con ES, con desarrollo de disnea en el 95 % de los pacientes.
De los 55 enfermos que se estudiaron, la mayoría tuvo disnea de esfuerzo, resultó ser el síntoma más frecuente. Un grupo importante estaba asintomático, este resultado nos confirma que el daño pulmonar puede no estar manifiesto en la clínica y obliga a la realización sistemática de PFR y TACAR.
La TACAR ha desplazado a la radiografía simple de tórax en el diagnóstico eficaz de la enfermedad intersticial. En los estadios tempranos el único hallazgo a encontrar es discreto vidrio esmerilado de predominio basal y subpleural, por lo que es importante solicitar este estudio en decúbito prono y supino, y así diferenciar entre real vidrio deslustrado y parénquima pulmonar comprimido.18
Los resultados mostraron un predominio del patrón en panal de abejas, seguido del de vidrio esmerilado, es más frecuente en nuestros pacientes la fibrosis pulmonar.
La presencia de vidrio deslustrado se correlaciona con la presencia de enfermedad reversible o “inflamación”, al menos que coexista y se encuentre con bronquiectasias por tracción, y con la presencia de infiltrado reticular y de panal de abeja, se correlaciona con la enfermedad irreversible o fibrosis.
Arbeláez Solera y otros12 plantean que la utilidad diagnóstica del estudio radica en la alta correlación entre el cuadro clínico y las pruebas de función respiratoria, en los pacientes con ES y EPI, cuando el compromiso pulmonar se evaluó con radiografía de tórax, su prevalencia es del 53 %; sin embargo, cuando se asoció con el TACAR fue mayor del 90 %, predominaron los pacientes con patrón en panal de abejas en un 74,3 %. Moazedi-Fuerst y otros11 señalan que la TACAR es el estudio de imagen estándar de oro en los pacientes con ES, en los cuales se evalúa compromiso respiratorio, pues la radiografía de tórax convencional no descarta compromiso pulmonar intersticial, es necesario este estudio en la evaluación inicial. Algunos estudios han demostrado que la TACAR tiene una sensibilidad del 95 % comparada con el 80 % de la radiografía convencional de tórax.19,20
En el estudio no se investigó la DLCO, se tuvo en cuenta que no es una determinación de fácil acceso en nuestro medio, pero si se pudo comprobar que la CVF fue reducida en la mayoría de los pacientes, lo que sugiere la presencia de un trastorno restrictivo, que está descrito como el más frecuente en los pacientes con EPI.
La PFR se utiliza durante el seguimiento de pacientes con EPI, dado que presenta alta predicción de la evolución de la enfermedad. Se demuestra un incremento de la mortalidad cuando los pacientes presentan una EPI restrictiva grave, definida por una capacidad vital forzada <50 %. En general la fibrosis pulmonar está caracterizada por presentar un patrón espirométrico restrictivo, con reducción de la CVF e incremento del cociente entre el volumen espiratorio forzado en el primer segundo (FEV1) y la CVF junto con una reducción de la DLCO.16
La variación significativa en la PFR, se considera que debe seguir las recomendaciones de la Sociedad Torácica Americana (ATS) y la Sociedad Europea de Enfermedades Respiratorias (ERS) para las neumopatías intersticiales. Así, la mejoría viene definida como un incremento ˃10 % en la CVF, el empeoramiento si existe un descenso ˂10 % y la estabilización si la variación no supera dichos extremos.21
La supervivencia libre de progresión (SLP) de la enfermedad, se define de manera similar a los ensayos clínicos de la fibrosis pulmonar idiopática: como el período de tiempo transcurrido hasta el punto en el que desciende la CVF al menos un 10 % o bien fallece el paciente. La supervivencia libre de enfermedad restrictiva grave se describe como el tiempo transcurrido hasta desarrollar una CVF inferior al 50 % o bien el fallecimiento.22
Durante el primer año tiene valor el descenso de la CVF o su variable combinada, y a los dos años de la enfermedad tiene más importancia el descenso de la difusión sobre la CVF por sí misma, lo cual parece indicar que la mortalidad en la EPI asociada a la ES no solo se debe a una evolución de la EPI sino a una progresión de la vasculopatía pulmonar no atribuible a la EPI.21,22
Existe evidencia sobre la importancia de los autoanticuerpos, tanto para el diagnóstico como para el pronóstico. En los trabajos sobre EPI asociada a la ES se ha observado que la evolución de dicha afección orgánica difiere según el anticuerpo presentado. En el grupo de pacientes estudiados el 80 % tuvo ANA positivo, y el 41,82 % anti-Scl-70. Los pacientes con ES presentan ANA positivos en el 90-100 % de los casos y no tienen correlación con la presencia o severidad del daño pulmonar. Los anticuerpos específicos que caracterizan a la enfermedad, si tienen fuerte asociación con las manifestaciones pulmonares y definen subgrupos clínicos de enfermos, por ejemplo, los anticuerpos anti-Scl-70, están presentes en el 40 % de los pacientes que tienen la forma difusa de la ES y se asocian con el desarrollo de alveolitis fibrosante. Generalmente los anticuerpos antitopoisomerasa 1 y anticentrómero son excluyentes entre sí.23
En el estudio los pacientes con anti-Scl-70 presentaron una mayor pérdida de la CVF, que traduce una mayor proporción de patrón restrictivo en los que tenían estos autoanticuerpos. Los resultados obtenidos demuestran significación estadística de las relaciones existentes entre estas variables.
El anticuerpo anti-Scl-70, se ha asociado a mayor agresividad en la enfermedad pulmonar.4
En el estudio de Doyle,3 expuso que los pacientes con anticuerpos anti-Scl-70 presentan un curso evolutivo de fibrosis más agresivo, con un deterioro rápido de la capacidad respiratoria pulmonar y un temprano desarrollo de enfermedad pulmonar restrictiva grave, con una supervivencia reducida.
Un estudio reciente realizado por Goh y otros24 se valora el poder predictivo de la evolución de las PFR en los 2 primeros años de la EPI sobre la mortalidad de esta. Dentro de los primeros 12 meses. Los factores asociados a mayor mortalidad son: el descenso ˂ 10 % en la CVF o bien un descenso moderado del 5-9 % junto con un descenso del 15 % en la DLCO.
Se concluye que el compromiso pulmonar y su relevancia como causa de morbimortalidad en la ES, es un hecho bien establecido. Esta afectación pulmonar puede no ser manifiesta clínicamente. El conjunto de pacientes con ES y EPID incluidos en el trabajo se caracterizó por una frecuencia de edad mayor de 40 años, del sexo femenino y pacientes de piel mestiza. Los pacientes presentaron de manera predominante la forma clínica difusa, con la disnea de esfuerzo como el síntoma más frecuente, y más de la mitad tuvo presente el fenómeno de Raynaud y tenía ANA positivo. La afectación pulmonar estuvo definida por el descenso de CVF en la espirometría y la presencia de afectación intersticial en las pruebas de imagen con patrón tomográfico en panal de abejas. La CVF disminuida, se asoció significativamente a la presencia de fenómeno de Raynaud, y a un comportamiento autoinmune positivo para anti-Scl-70.

