










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Pediatría
On-line version ISSN 1561-3119
Rev Cubana Pediatr vol.71 no.4 Ciudad de la Habana Oct.-Dec. 1999
Experiencia y resultados
Hospital Pediátrico Docente "William Soler", Ciudad de La HabanaExpresión fenotípica de una sordera familiar con deleción del gen POU3F4
Dra. Ibis Menéndez,1 Dra. Manuela Villamar,2 Dra. Blanca Carrillo,1 Dr. Ignacio del Castillo,2 Téc. Lourdes Romero,2 Dra. Maribel Ponce de León1 y Dr. Felipe Moreno2RESUMEN
Se presenta una familia cubana con 5 miembros afectados por una hipoacusia bilateral, congénita, severa, mixta con componente neurosensorial predominante y sin alteraciones morfológicas de oído interno. El patrón de transmisión era compatible con la herencia recesiva ligada al cromosoma X. Los estudios moleculares detectaron una deleción en la región Xq21.1 que implica el gen POU3F4, responsable de la sordera de tipo DFN3. Se hacen comentarios sobre la evidente variabilidad clínica de las sorderas tipo DFN3.Descriptores DeCS: PERDIDA AUDITIVA BILATERAL/genética; PERDIDA AUDITIVA BILATERAL/etiología; TRASTORNOS DEL LENGUAJE/etiología; PERDIDA AUDITIVA BILATERAL/complicaciones; FONDO DE OJO.
Las sorderas ligadas al cromosoma X representan entre el 1 al 5 % de las sorderas hereditarias no sindrómicas.1 Son heterogéneas, tanto desde el punto de vista clinicoaudiológico (congénitas o progresivas; neurosensoriales, conductivas o mixtas) como del genético con 4 loci actualmente mapeados.2-4 La forma más frecuente de sordera ligada al cromosoma X, el tipo DFN3, se describió inicialmente en individuos con características clinicoaudiológicas bien definidas (MIM 304400): sordera mixta progresiva con fijación del estapedio y "gusher" de perilinfa durante la estapedectomía.5,6 Esta sintomatología tan característica facilitó el abordaje desde el punto de vista molecular. Sin embargo, se han descrito deleciones del gen POU3F4 en pacientes con reordenamientos cromosómicos del brazo largo del cromosoma X;7-9 mutaciones puntuales del gen POU3F4 en pacientes con hipoacusias neurosensoriales o mixtas asociadas con anomalías de oído interno con "gusher" o sin éste;10 por último se han descrito pacientes con sordera tipo DFN3 en los cuales el gen POU3F4 está presente y sin alteración de su secuencia.10,11
En este trabajo se reportan los hallazgos clínicos y genético-moleculares en una familia cubana con una sordera congénita severa que afecta sólo a varones y se hacen algunas consideraciones sobre la evidente variabilidad clínica interfamiliar de las DFN3.
MÉTODOS
Investigaciones clínicas
Se estudiaron 19 individuos que pertenecían a la familia, de ellos, 5 afectados con una sordera congénita severa con afectación del lenguaje. A los hipoacúsicos se les realizó examen de fondo de ojo, investigaciones endocrinas (determinación de niveles de FSH, LH y testosterona basal), tests psicométricos y tomografía axial computadorizada (TAC) de oído interno. Se realizaron investigaciones citogenéticas en los individuos III:11, III:13, IV:3 y IV:6 (2 portadoras obligadas, un afectado y un varón normal respectivamente).Investigaciones genético-moleculares
Se obtuvo ácido desoxirribonucleico (DNA) de sangre periférica de los 19 individuos por métodos estándares.13 Se realizaron amplificaciones PCR con un termociclador Perkin-Elmer 9 600 de los siguientes marcadores microsatélites: DXS1225, DXS995, DXS8076, otros marcadores no microsatélites como el DXS169, DXS26, DXS232, DXS355, DXS326 y las STS 71:3, 24:17, 34:2, y 24:5 en todos los miembros de la familia. Las condiciones de amplificación fueron las descritas por Robinson y otros y Dahl y otros.14,15Para determinar la presencia/ausencia del gen POU3F4 se amplificó la ORF completa del gen en todos los varones afectados y se visualizó en geles de agarosa; en las portadoras obligadas y posibles éste se determinó mediante hibridación, con la utilización como sonda del producto amplificado del gen completo marcado con 32P.
RESULTADOS
Al examen físico no se apreciaron síntomas o signos acompañantes de la sordera. Los resultados de las investigaciones que se realizaron para descartar coroideremia, anomalías cromosómicas, anomalías de oído interno, endocrinopatías y retraso mental fueron normales.Características de la pérdida auditiva
El estudio audiométrico en los 5 afectados destacó una hipoacusia bilateral severa que afectó a todas las frecuencias, con reflejo estapedial abolido y gran reserva coclear. En el timpanograma se observó una marcada disminución de la compliancia en ambos oídos, con la consiguiente disminución de la movilidad de las membranas timpánicas. En algunas hembras el reflejo estapedial estaba abolido, sin afectación de la audición.Estudio genético-molecular
El patrón de transmisión de la sordera era característico de la herencia recesiva ligada al cromosoma X (fig.1).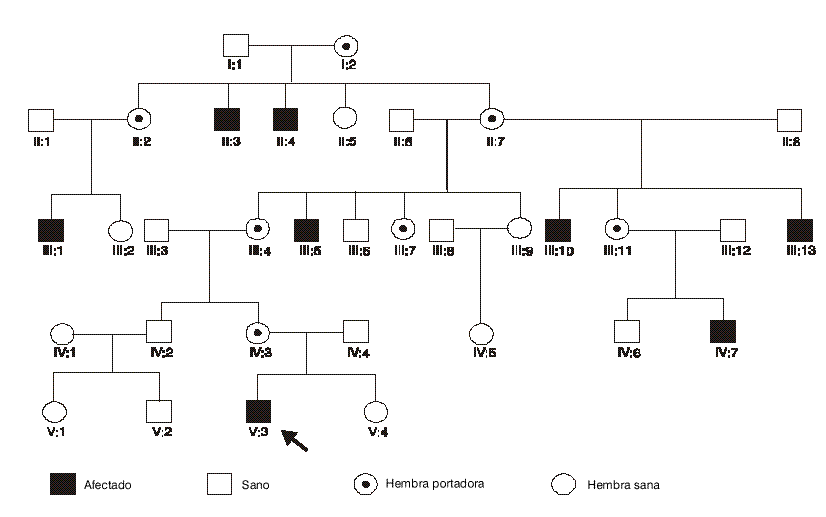
FIG. 1. Árbol genealógico de la familia.
Los afectados de esta familia mostraron una deleción delimitada por los marcadores DXS26 y DXS232, ambos presentes y flanqueantes al gen POU3F4; de los marcadores microsatélites DXS995 y DXS8076, así como de la ORF completa del gen POU3F4 no se obtuvieron productos amplificados (fig. 2). El estado de portador se determinó por análisis de Southern blot (análisis genético directo) que permitió comprobar la presencia del gen en doble dosis en las hembras sanas y en monodosis en las hembras portadoras. Simultáneamente se configuró el haplotipo para los marcadores microsatélites DXS1225, DXS995, DXS8076 y CHM (análisis genético indirecto).
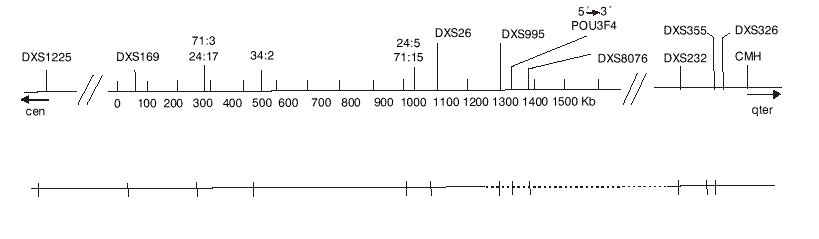
FIG. 2. Mapa físico de la región crítica del locus DFN3. En la parte superior se señala la localización del gen POU3F4 y de los marcadores utilizados en el estudio. En la parte inferior se muestra con líneas de puntos la deleción detectada en los pacientes. Las rayas verticales corresponden a la posición de los marcadores.
DISCUSIÓN
La pérdida auditiva fue bastante homogénea en todos los afectados. El componente neurosensorial predominó y enmascaró el conductivo. No se encontraron anomalías del oído interno, ni se ha verificado progresión de la sordera en las evaluaciones realizadas; tampoco se observó la presencia de otros signos o síntomas que hicieran sospechar del tipo de sordera DFN3, con excepción de la herencia ligada al cromosoma X. Es de destacar el hallazgo de reflejo estapedial abolido, sin pérdida auditiva, en todas las portadoras obligadas confirmadas molecularmente, lo cual será motivo de un estudio ulterior.Los estudios moleculares se iniciaron investigando el locus DFN3, porque las mutaciones en esta región explican aproximadamente el 50 % de las hipoacusias ligadas al cromosoma X.7,10-12,14 Deleciones de la región Xq21.1 como la encontrada en esta familia son el tipo de mutación más frecuente entre las sorderas DFN3.7,10,11
La asociación entre sordera de tipo DFN3 y mutaciones puntuales del gen POU3F4 fue descrita por de Kok y otros.10 Bitner Glindzin y otros encontraron deleciones del gen POU3F4 en sorderas neurosensoriales con anomalía ósea de oído interno, que ampliaron el espectro clínico de las DFN3,11 y nosotros la reportamos en esta sordera no sindrómica, bilateral, congénita, severa, conductivo-neurosensorial sin anomalías tomográficas demostrables del oído interno. En general entre las sorderas tipo DFN3 se observan diferencias importantes en relación con el tipo de hipoacusia, anomalías morfológicas del oído interno y la edad de comienzo de la pérdida auditiva,7,10-12,16 sin que se haya podido establecer una estricta correlación genotipo-fenotipo por el momento. En ausencia de "gusher", la sordera DFN3 suele sospecharse en un paciente masculino con hipoacusia y malformaciones del oído interno tomográficamente detectadas, o con coroideremia, o retraso mental o con alteraciones cromosómicas que incluyan a la región Xq21.1. Nuestra experiencia con esta familia sugiere comenzar los estudios moleculares en las sorderas ligadas al cromosoma X por el locus DFN3, porque son las más frecuentes y además muy heterogéneas clínicamente.
Aunque Cremers y otros han hallado pacientes DFN3 en los que el gen POU3F4 está intacto,10,12,16 está claramente demostrado que las mutaciones del gen POU3F4 y/o de sus zonas adyacentes causan sorderas.10,12,16 En un futuro próximo las investigaciones en las DFN3 deberán centrarse en identificar a los genes que regulan este factor transcripcional. Ello debe contribuir a la explicación de la variabilidad clínica de las DFN3.
AGRADECIMIENTOS
Los autores desean dejar constancia de su agradecimiento al programa Fondo de Expertos de la AECI y a la Consejería de Educación y Cultura de la Comunidad de Madrid, por financiar este trabajo, así como a los miembros de la familia V.T. por su extraordinaria colaboración en las investigaciones realizadas.SUMMARY
A Cuban five-member family affected by a severe congenital bilateral mixed deafness with predominant sensorineural component and without morphological changes in the internal hearing is presented in this study. The transmission pattern was compatible with X-linked recessive heritage. The molecular studies detected a deletion of Xq 21 region involving POU3F4 gene which is responsible for DFN3-type deafness. Comments are made on the obvious clinical variability of DFN3-type deafness.Subject headings: HEARING LOSS, BILATERAL/genetic; HEARING LOSS, BILATERAL/etiology; HEARING LOSS, BILATERAL/complications; LANGUAGE DISORDERS/etiology; FUNDUS OCULI.
REFERENCIAS BIBLIOGRÁFICAS
- Cohen MM, Gorlin JJ. Epidemiology, etiology and genetic patherns. En: Gorlin RJ, Torriello HV, Cohen MM, eds. Hereditary hearing loss and its syndromes. New York: Oxford University Press, 1995:9-21.
- Van Camp G, Willens PJ, Smith RJH. Nonsyndrome hearing impairment: Unparalleled heterogeneity. Am J Hum Genet 1997;60:758-64.
- Petit C. Genes responsible for human hereditary deafines: Symphony of a thousand. Nature Genet 1996;14:385-91.
- Castillo I del, Villamar M,Sarduy M, Romero L, Herraiz C, et al. A novellocus for non-syndromic sensorineural deafines (DF N6) maps to chromosome Xp22. Hum Mol Genet 1996;5:1383-7.
- Mac Kusick VA. X linked Phenotypes. En: Mendelian Inheritance in Man. Catalogs of dominant autosomal recesive, and X-linked phenotypes. 9th ed. edition Baltimore: Johns Hopkins University Press; 1990;1584.
- Nance WE, Seleff R, McLeod A, Sweeney A, Cooper C, McConnell F. X linked mixed deafiness with congenital fixation of the stapedial footplate and perilymphatic gusther. Birth defects 1971;4:64-9.
- Huber I, Bitner-Glindzicz M, Kok YTM de, Maarel SM, van der, Ischikawa-Brush Y, et al. X linked mixed deafines (DFN3). Cloning and characterization of the critical region allows the identification of novel microdeletions. Hum Mol Genet 1994;3:1151-4.
- May M, Colleaux L, Murgie A, Aylswork A, Nussbaum R, et al. Molecular analysis of four males with mental retardation and deletions of Xq21 places the putative MR region in Xq21.1 between DXS233 and CHM. Hum Mol Genet 1995;4:1465-6.
- Maarel SM van der, Scholten IHJ, Maat-Kievit JA, Hubert I, Kok YM de, et al. Yeast artificial chromosome cloning of the Xq13.3-q21.31 region and fine mapping of a deletion associated with choroideremia and nospecific mental retardation. Eur J Hum Genet 1995;3:207-18.
- Kok YJM de, Maarel SM van der, Bitner-Glinzicz M, Huber I, Monaco AP, et al. Association between X-mixed deafiness and mutations in the POU domain gene POU3F4. Science 1995;267:685-8.
- Bither Glindzicz M, Tumpenny P, Hoglund P. Kaareainen H, Sankila EM, et al. Further mutations in Brain 4 (POU3F4) clarity the phenotype in the X-linked deafiness, DFN3. Hum Mol Genet 1995;4:1467-9.
- Kok YJM de, Merkc G, Maarel SM van der, Huber I, Malcom S, et al. A duplication/paracentric inversion associated with familial X-linked deafiness (DFN3) suggest the presence of a regulatory element more than 400kb upstream of the POU3F4 gene. Hum Mol Genet 1995;4:2145-50.
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 1988;16:1215.
- Robinson D, Lamont M, Curtis G, Shields DC, Phelps P. A family with X-linked deafines showing linkage to the proximal Xq region of the X chromosome. Hum Genet 1992;90:316-8.
- Dahl N, Laporte J, Hu I, Biancalana V, LePasttier D, Cohen D, et al. Deletion mapping of X linked mixed deafines (DFN3) identifies a 265-525 kb region centromere of DX526. Am J Hum Genet 1995;56:999-1001.
- Friedman RA, Byknoskaya MS, Tu G, Talbot MJ, Wilson DF, et al. Molecular analysis of the POU3F4 gene in patients with clinical and evidence of X linked mixed deafines with perilymphatic gusther. Am Othol Rhinol Laryngol 1997;106:320-25.
Recibido: 15 de febrero de 1998. Aprobado: 12 de febrero de 1999.
Dra. Ibis Menéndez. Hospital Pediátrico Docente "William Soler", 100 y Perla, municipio Boyeros, Ciudad de La Habana, Cuba.
2 Unidad de Genética Molecular. Hospital "Ramón y Cajal", Madrid, España.

