










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Pediatría
On-line version ISSN 1561-3119
Rev Cubana Pediatr vol.80 no.3 Ciudad de la Habana July-Sept. 2008
PRESENTACIÓN DE CASOS
Presentación de un caso de hemidisplasia congénita con ictiosis eritrodérmica
Report of a congenital hemidysplasia with erythrodermic ichthyosis
Andrés A. Morilla Guzmán,I Norma Elena de León Ojeda,II Aida Elena García del Collado,III Alida Petizco Hernández,IV Tania Rodríguez Suárez,V Sonia Correa Santos,VI Maisbell Rodríguez Fuertes,VII Silvia Miyares CaoVIII
I Especialista de I Grado en Neonatología. Asistente. Hospital Pediátrico "Angel A. Aballí". La Habana, Cuba.
II Especialista de II Grado en Genética Clínica. Asistente. Hospital Pediátrico "William Soler". La Habana, Cuba.
III Especialista de I Grado en Neonatología. Instructor. Hospital General "Enrique Cabrera". La Habana, Cuba.
IV Especialista de I Grado en Neonatología. Hospital General "Enrique Cabrera". La Habana, Cuba.
V Residente de Neonatología. Hospital Pediátrico "Angel A. Aballí". La Habana, Cuba.
VI Residente Materno Infantil. Hospital General "Enrique Cabrera". La Habana, Cuba.
VII Residente de Neonatología. Hospital General "Enrique Cabrera". La Habana, Cuba.
VIII Especialista de I Grado en Dermatología. Hospital Pediátrico "William Soler". La Habana, Cuba.
RESUMEN
La hemidisplasia congénita con ictiosis y defectos de las extremidades es una enfermedad infrecuente, hereditaria, monogénica, que se transmite como un rasgo dominante ligado al cromosoma X. Se presenta el caso de una paciente con este diagnóstico clínico neonatal, que presentaba eritrodermia ictiosiforme en el hemicuerpo derecho, acompañada de hipomelia del miembro superior e inferior derechos, defectos óseos en miembros afectados y columna vertebral, agenesia renal unilateral, cardiopatía congénita de tipo comunicación interventricular conoventricular y arteria umbilical única. Se realizaron las interconsultas necesarias, estudios sonográficos y radiológicos para completar el diagnóstico y se ofreció asesoramiento genético y seguimiento del caso según las complicaciones reportadas en la literatura médica y los hallazgos clínicos de la paciente.
Palabras clave: Síndrome CHILD, recién nacido.
ABSTRACT
Congenital hemidysplasia with icthyosis and limb defects is a hereditary, monogenic and infrequent disease transmitted as a dominant trait linked to the X chromosome. The case of a female patient with this neonatal clinical diagnosis showing ichthyosiform erythroderma on the right hemibody, accompanied with hypomelia of the right upper and lower limbs, bone defects in the affected limbs and spinal column, unilateral renal agenesia, congenital heart disease with inter- and conoventricular communication, and a unique umbilical artery was reported. The necessary inter-consultations were arranged and sonographic and radiological studies were conducted to complete the diagnosis. Genetic counselling was given and the case was followed up according to the complications reported in medical literature, and to the clinical findings of the patient.
Key words: CHILD'S syndrome, newborn.
INTRODUCCIÓN
El síndrome CHILD tiene otras sinonimias como eritrodermia ictiosiforme unilateral, con malformaciones ipsolaterales de las extremidades; hemidisplasia congénita con ictiosis eritrodérmica y defectos de las extremidades; hemidisplasia unilateral, y alteración de la cornificación tipo 16. Su nombre se corresponde con las siglas en inglés de hemidisplasia congénita con ictiosis y defectos de las extremidades (Congenital Hemidysplasia with Ichthyosiform erythroderma and Limb Defects).1
Se creía que un informe de Zellweger y Uehlinger, de 1948, representaba el primer caso publicado del síndrome de CHILD; sin embargo, se ha encontrado un informe más antiguo, publicado en 1903 por Otto Sachs.2 No obstante, el primer caso que reconoce la literatura genética es el de 2 hermanos, publicado por Falek en 1968 y Shear en 1971, pero fue Happle, en 1980, quien usó por primera vez el acrónimo.3
Se considera una enfermedad rara, hereditaria, monogénica, que se transmite como un rasgo dominante ligado al cromosoma X; es letal en varones, y en la mayoría de los casos está causado por mutaciones en el gen NSDHL (NADPH proteína análoga de la deshidrogenasa esteroidea), localizado en el locus Xq28 e implicado en el metabolismo del colesterol.1-7 Los que describen el síndrome de CHILD de manera atípica, con el nevo en el lado izquierdo o distribuido de manera bilateral, lo asocian con otras mutaciones en el mismo gen.8 Otros casos tienen mutaciones en el gen EBP, descrito como causal de la condrodisplasia punctata.3
La proporción de sexos es de 1 varón por cada 28 hembras, ya que la mayoría de los fetos masculinos que se afectan no llegan al término de la gestación; el lado más frecuentemente afectado es el derecho9, en una proporción de 7-31, otros describen una proporción de 14-6.10
Las manifestaciones clínicas que distinguen a este síndrome son las características dermatológicas y las alteraciones de los miembros del mismo lado. En la literatura5 describen las manifestaciones dermatológicas como el nevo CHILD, refiriéndose al nevo epidérmico inflamatorio e ictiosiforme de disposición unilateral; otros1,4 lo describen como un eritema unilateral y escalado, con demarcación distinguible en medio del torso. Esta dermatosis puede presentarse al nacimiento o se puede desarrollar durante las primeras semanas de vida.
Las alteraciones ipsolaterales asociadas pueden ser variadas, desde una hipoplasia en diferentes dedos, hasta la ausencia completa de una extremidad. Pueden encontrarse también hipoplasia en el mismo hemicuerpo de otras estructuras esqueléticas, defectos en el cerebro y en otros órganos internos.4,6,7
Las otras anomalías asociadas a esta entidad pueden ser: retraso del crecimiento intrauterino, contracturas articulares, pterigium en codos y rodillas, alopecia unilateral e hiperqueratosis, displasia ungueal, hipoplasia de mandíbula, clavícula, escápula, costillas y vértebras; escoliosis; alteraciones cardiovasculares, como comunicación interauricular e interventricular, ventrículo único y agenesia renal unilateral, y leve retraso mental. Con menos frecuencia se han descrito: hipoplasia unilateral de cerebro, nervios craneales, médula espinal, pulmones, tiroides, suprarrenales, ovario y trompas de Falopio. Se ha visto asociado también, de manera infrecuente, labio leporino, hernia umbilical, sordera, hidrocefalia y mielomeningocele.1
PRESENTACIÓN DEL CASO
Recién nacida, de la raza blanca y sexo femenino, hija de padres no consanguíneos. Madre de 27 años de edad, con antecedentes de hernia hiatal, infección vaginal por Monilia durante el embarazo, para lo que llevó tratamiento local, y sangramiento durante el 1er. trimestre del embarazo; el padre, de 40 años de edad, no tiene antecedentes familiares ni personales relevantes.
Nace producto de una cesárea realizada por presentación pelviana, con rotura prematura de membranas (RPM), a las 38 semanas de edad gestacional, con líquido amniótico claro y una circular laxa al cuello; puntuación del Apgar 9/9 y peso de 2 970 g, con una valoración nutricional entre un 25 y 50 percentil, según tablas cubanas.11
Desde el momento del nacimiento se aprecian las características clínicas externas que se describen en el paciente. Al examen físico se observa piel en hemicuerpo izquierdo de aspecto, textura y color normal; en hemicuerpo derecho, piel seca y descamada, de aspecto ictiosiforme y coloración eritematosa, bien delimitada en línea media longitudinal del cuerpo, con excepción de la cabeza. Ausencia de lanugo en hemicuerpo afectado (figura 1). Cráneo aparentemente normal. Cara redonda y plana, con puente nasal deprimido y epicanto bilateral. Columna vertebral con desviación de concavidad derecha.
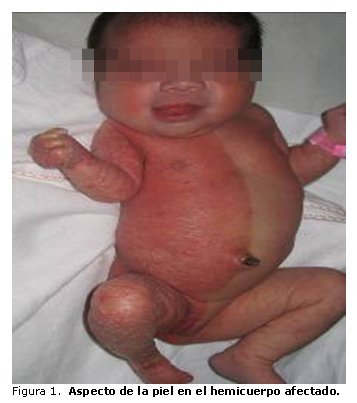
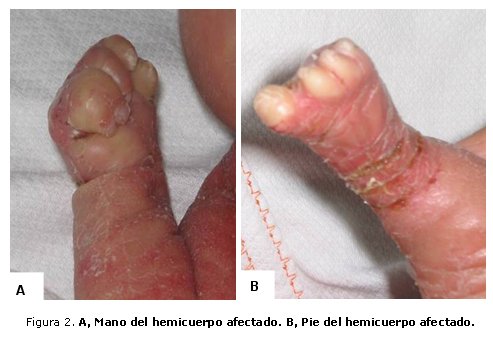
En las extremidades se observa hipomelia (acortamiento) de los miembros superior e inferior derechos. Mano derecha con rigidez articular de la muñeca; los 5 dedos en flexión con hipoplasia ungueal y sindactilia membranosa del 4to. y 5to. dedo (figura 2 A). Pie derecho atrófico, con los 5 dedos presentes con hipoplasia ungueal (figura 2 B). Miembros superior e inferior izquierdos normales; muñón umbilical con arteria umbilical única.
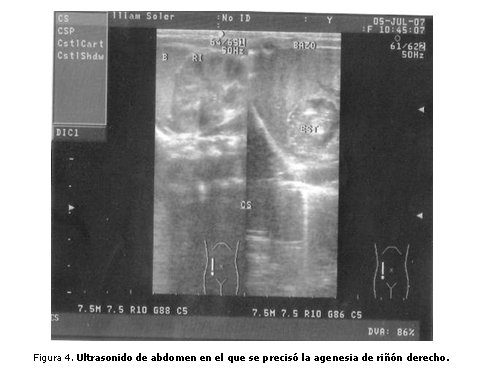
En el aparato cardiovascular se percibe soplo sistólico audible en borde esternal izquierdo II/VI. Complementarios: se realiza rayos X de tórax-abdomen, en el que se aprecia escoliosis y hemivértebras 7ma. y 9na. torácicas (figura 3). Ultrasonido de cráneo transfontanelar, con resultado normal. Ultrasonido de abdomen que revela agenesia de riñón derecho (figura 4). Ecocardiograma que manifiesta comunicación interventricular (CIV) conoventricular subaórtico de 5 mm.

DISCUSIÓN
El síndrome CHILD es una enfermedad infrecuente, que se ha insertado dentro del grupo de los mosaicismos funcionales del cromosoma X, en los fenotipos no letales12 y constituye un síndrome por la presencia de múltiples defectos congénitos malformativos, de etiología genética.13
Se dice que este llamativo síndrome pudiera ser una variedad del síndrome de Conradi Hunermann; ambos tienen similitudes clínicas y bioquímicas, con el aumento de 8 dihidrocolesterol, pero se diferencian en su localización, generalizada en uno y parcial en el otro, incluso, el gen EBP se ha encontrado mutado en casos con síndrome CHILD y causa también el Conradi3, y ambos son dominantes, ligados al cromosoma X.13
El diagnóstico prenatal resulta difícil y solo pudiera ser sospechado por la presencia de la cardiopatía congénita y la agenesia renal, además del hipocrecimiento; aún así, por su infrecuencia, sería difícil sospecharlo. Su diagnóstico posnatal se realiza al nacer, por parte de los neonatólogos que asisten al parto, aunque puede efectuarse posteriormente, cuando las manifestaciones clínicas no son evidentes en la primera semana de vida.9 Se recomienda la valoración con genetistas clínicos que, como en este caso, avalen el diagnóstico y conduzcan el seguimiento con diferentes especialidades.
El caso que se presenta reúne muchas de las manifestaciones clínicas descritas en la literatura y se acompaña de las anomalías de órganos referidas, como la escoliosis, las malformaciones vertebrales, la agenesia renal ipsolateral y la comunicación interventricular. Llama la atención en este caso, la presencia de arteria umbilical única, que resulta un hallazgo poco frecuente y se presenta en pocas entidades genéticas, como la asociación VATER10, mas no ha sido reportada en este síndrome.
El diagnóstico clínico, según los hallazgos en la paciente, coinciden con lo reportado por König A y colaboradores, que aseguran que el diagnóstico puede ser clínico, si se tiene en cuenta que las características morfológicas del nevo CHILD son propias de esta entidad, y la distribución simétrica del mismo es excepcional.7 No obstante, se han descrito mutaciones en el NSDHL (Xq28) y en el EBP (Xp11.2); de poder realizarse el estudio molecular, solo confirmaría la mutación causante, mas no variaría el asesoramiento genético, pues ambas son ligadas al cromosoma X, dominantes.
La evolución clínica de esta enfermedad, descrita en otros casos, refiere desarrollo de las lesiones de piel que pueden incluir nuevas áreas. Este caso tiene una comunicación interventricular pequeña, sin repercusión hemodinámica, que puede tener un seguimiento ambulatorio, al igual que la función renal.
El tratamiento de esta entidad se orientará hacia el asesoramiento genético y el seguimiento por genetistas, apoyo psicológico a la familia, el seguimiento continuo de su cardiopatía por la red cardiopediátrica de su área de salud, y la conducta dermatológica para mejorar las lesiones de piel que se presentan en este síndrome.
REFERENCIAS BIBLIOGRÁFICAS
1. Izquierdo M, Avellaneda A, editores. CHILD, Síndrome. España [monografía en internet]. 2003 [citado 2007 jul 13]. Disponible en: http://iier.isciii.es/er/prg/er_bus2.asp cod_enf1390.html 2. Bittar M, Happle R. CHILD syndrome avant la lettre. J Am Acad Dermatol. 2004;50(2 Suppl):S 34-7.
3. On Line mendelian Inheritance in man [citado 2007 ago 1]. Disponible en: http://www.ncbi.nlm.nih.gov/OMIM/
4. Web médica argentina. Guía de Síndromes. Materias [homepage en internet]. Argentina: Materias [citado 2007 jul 13]. Disponible en: http://www.webmedicaargentina.com.ar/MATERIAS/sindromes1
5. Iqb, editor. Genes implicados en enfermedades. España [monografía en internet] [citado 2007 jul 13]. Disponible en: http://www.iqb.es/diccio/g/gen.htm
6. Fink-Puches R. Orpha, editor. CHILD, Síndrome. España [monografía en internet]. 2007 [citado 2007 jul 13]. Disponible en: http://www.orpha.net/static/es/child.html.
7. König A, Happle R, Fink-Puches R, Soyer HP, Bornholdt D, Engel H, et al. A novel missense mutation of NSDHL in an unusual case of CHILD syndrome showing bilateral, almost symmetric involvement. J Am Acad Dermatol. 2002;46(4):594-6.
8. Hummel M, Cunningham D, Mullett CJ, Kelley RI, Herman GE. Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. Am J Med Genet. 2003;122(3):246-51.
9. Kim CA, Konig A, Bertola DR, Albano LM, Gattás GJ, Bornholdt D, et al. CHILD syndrome caused by a deletion of exons 6-8 of the NSDHL gene. Dermatology. 2005;211(2):155-8.
10. Jones KL. Smith's Recognizable Patterns of Human Malformation. 6th Ed. Montreal: Elsevier,2006.
11. Valdés R, Reyes DM. Examen Clínico al Recién Nacido. La Habana: Editorial Ciencias Médicas,2004.
12. Sisbib M, Rudolt H. Mosaicismos Cutáneo: Concepto y Consecuencias Clínicas. Dermatología Peruana. Edición Especial. [serie en internet]. 2000 sept [citado 2007 jul 13] [aprox. 2 p.]. Disponible en: http://sisbibunmsm.edu.pe/BVRevistas/dermatologia/es/mos_cut.htm
13. Siegfried EC, Esterly NB. Enfermedades congénitas y hereditarias de la piel. En: Avery ME. Tratado de neonatología de Avery, 7ma. ed. España: Harcourt Saunders; 2001. Pp. 1282-96.
Recibido: 10 de marzo de 2008.
Aprobado: 23 de julio de 2008.
Andrés A. Morilla Guzmán. Calle Julio A. Mella Edif. No.5 Apto 36 e / 1era y 2da. Rpto. Eléctrico Municipio Arroyo Naranjo. La Habana, Cuba. Correo electrónico: andres.morilla@infomed.sld.cu

