










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Pediatría
On-line version ISSN 1561-3119
Rev Cubana Pediatr vol.88 no.1 Ciudad de la Habana Mar.-Mar. 2016
Rev Cubana Pediatr. 2016;88(1)
PRESENTACIÓN DE CASO
Deficiencia congénita de proteína C en un recién nacido con trombosis y necrosis de tejidos extensa
Congenital C protein deficiency in a newborn with extensive thrombosis and necrosis of tissues
Dra. Bárbara Acosta Batista, Dr. Manuel Díaz Álvarez, Dra. Raquel Fernández Nodarse, Dra. Libertad Rivera Alés, Dr. Orestes Chagues Leyva
Servicio de Neonatología. Hospital Pediátrico Universitario “Juan Manuel Márquez”. La Habana, Cuba.
RESUMEN
Uno de los trastornos hematológicos más graves del período neonatal es la deficiencia congénita de proteína C, de presentación muy rara, y causa de enfermedad tromboembólica severa y púrpura fulminante en recién nacidos. Se puede sintetizar como una entidad clínico-patológica, de aparición aguda, con trombosis de la vasculatura de la dermis, lo cual conduce a necrosis hemorrágica y progresiva de la piel, asociada a coagulación intravascular diseminada y hemorragia perivascular, que ocurre en el período neonatal. El paciente presentado exhibe los elementos clínico-patológicos que caracterizan la púrpura fulminante, cuyo origen se debe a una deficiencia hereditaria de proteína C, lo cual condujo a la aparición de complicaciones trombóticas severas.
Palabras clave: púrpura fulminante, tromboembolismo, deficiencia de proteína C hereditaria, coagulación intravascular diseminada, recién nacido.
ABSTRACT
One of the most serious hematological disorders of the neonatal period is congenital C protein deficiency of very rare occurrence and the main cause of severe thromboembolic disease and purpura fulminans in newborns. It may be summarized as a clinical and pathological entity of acute occurrence, with dermis vasculature thrombosis that leads to progressive hemorrhagic necrosis of the skin, associated to disseminate intravascular coagulation and perivascular hemorrhage in the neonatal period. The patient of this report showed the clinical and pathological elements characterizing purpura fulminans the origin of which is due to hereditary C protein deficiency that led to onset of severe thrombotic complications in this patient.
Keywords: purpura fulminans, thromboembolism, hereditary C protein deficiency, disseminate intravascular coagulation, newborn.
INTRODUCCIÓN
El tromboembolismo venoso en la infancia es una enfermedad rara; sin embargo, está recibiendo una mayor atención para su diagnóstico y seguimiento. La trombofilia es definida como un desorden de la hemostasia, con tendencia a la aparición de trombosis venosa o arterial, debido a anomalías en la composición de la sangre, flujo sanguíneo o la pared vascular, y el término indica la naturaleza hereditaria de la enfermedad.1 Es más a menudo usado para la trombosis venosa, que es considerada como una enfermedad separada de la arterial, ya que su patogénesis es muy diferente.
Uno de los trastornos hematológicos más graves del período neonatal es la purpura fulminans o púrpura fulminante (PF), que puede presentarse en el curso de infecciones bacterianas severas, como sepsis y meningitis, en que se asocia a consumo de proteínas C y S;2-5 así como también asociada a deficiencia congénita de proteínas C y S.6 Se considera como una entidad clínico-patológica, de aparición aguda, con trombosis de la vasculatura de la dermis, lo cual conduce a necrosis hemorrágica y progresiva de la piel, asociada a coagulación intravascular diseminada y hemorragia perivascular, que ocurre en el período neonatal. Tiene una evolución rápida y dramática si no se instaura un tratamiento adecuado. Se presenta con lesiones purpúricas, equimóticas difusas y simétricas, que evolucionan en pocas horas a bulas hemorrágicas y/o extensas áreas de necrosis, comprometiendo, generalmente, áreas distales bilaterales y simétricas y de trauma o presión, como son los glúteos, los talones, la zona sacra y el cuero cabelludo.5-8 Sin tratamiento oportuno, estos pacientes evolucionan a eventos trombóticos mayores con compromiso neurológico, oftalmológico, pulmonar y también amputación de miembros.7-12
A propósito de haber asistido a un neonato con esta afección y su dramática evolución, con complicaciones severas de trombosis y necrosis extensa, es que decidimos exponer este caso.
PRESENTACIÓN DEL CASO
Neonato del sexo femenino, de 12 días de edad, que se traslada a nuestro servicio de Neonatología para ser evaluado por las especialidades pediátricas de Hematología, Cirugía y Caumatología, al presentar lesiones purpúrico-necróticas en varias localizaciones del cuerpo.
La historia del hospital de referencia describe una madre de 21 años de edad, G2P1A1 (provocado), con antecedentes de salud anterior y embarazo de curso normal, no consanguinidad, ni enfermedades hematológicas familiares. El parto fue a las 41 semanas de gestación, distócico por cesárea, debido a desproporción céfalo-pélvica, extracción difícil y demorada, con peso al nacer de 3 300 g, y puntaje de Apgar 1-3-8. Se administró profilaxis con vitamina K.
Recibió medidas de reanimación cardiopulmonar y ventilación durante 78 h, tuvo cateterismo umbilical por 72 h, acceso venoso percutáneo y recibió antibioticoterapia. Antes de las 48 h de nacido presentó lesiones purpúricas en miembros inferiores con equímosis y hematomas, las que se acentuaron en los días siguientes con nuevas lesiones en el pie y la pierna derecha en forma de bota, muslo y glúteo izquierdo, que alcanzaba los genitales, de iguales características, con grandes hematomas que presentaban flictenas. Los exámenes de perfil de sepsis iniciales (leucograma y proteína C reactiva) fueron normales, aunque la hemoglobina presentó un descenso importante, por lo que requirió transfusión de glóbulos y hemocultivo de ingreso sin aislamiento bacteriano.
Se recibe en este servicio de Neonatología con mala apariencia, palidez generalizada y pobre vitalidad. Al examen físico se constatan trastornos hemodinámicos, y se ingresa de inmediato en cuidados intensivos. Se comenzó la reposición de volemia, drogas vasoactivas y antibióticos de amplio espectro (vancomicina, ceftazidime y metronidazol).
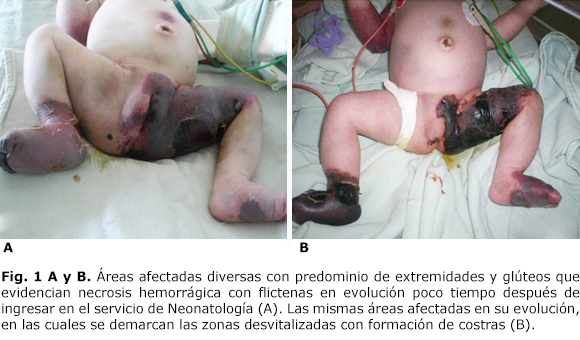
Se verifica la presencia de hematomas en distintas localizaciones de una profundidad que abarca todos los tejidos blandos y con variable compromiso tisular (hematoma supra-umbilical redondeado), gran hematoma que abarcaba todo el muslo, el glúteo izquierdo y hasta los labios mayores en la región genital; además, de manera aislada, la planta del pie del miembro inferior de ese mismo lado, en miembro inferior derecho similares lesiones que ocupaban todo el pie y tercio distal en forma de bota, en la región antero-interna del codo derecho y en el cuero cabelludo de la región parieto-occipital derecha. La piel en estas localizaciones estaba de color rojizo en vino de oporto, algunas con zonas violáceas a negruzcas con flictenas debridadas y partes de tejido desvitalizado (figura 1A).
Los exámenes complementarios al ingreso mostraron lámina periférica con conteo de leucocitos normal, macroplaquetas; proteína C reactiva positiva, velocidad de sedimentación globular 5 mm/h. El coagulograma mostró tiempo de coagulación 25', el conteo de plaquetas fue 80 x 109/L, el tiempo de protrombina 17'' (control 14''), el tiempo parcial de tromboplastina fue 1' 40'' (control 24-36''), coágulo irretráctil, dímero D positivo. Se indicó transfusión de plasma fresco y concentrado de plaquetas. En examen ultrasonográfico de cabeza y abdomen no se visualiza hematoma o sangrado intracraneal, ni intraabdominal.
El paciente fue discutido en el servicio de Neonatología, en conjunto con las especialidades de Hematología, Angiología, Ortopedia, Cirugía Reconstructiva y Caumatología, y se planteó el diagnóstico probable de PF. Se mantuvo durante los primeros días con transfusiones de plasma fresco y concentrado de plaquetas, y al cuarto día de esta terapia se normaliza coagulograma. Posteriormente, se estableció un régimen de transfusiones diarias de plasma fresco, con monitoreo de coagulograma que se mantuvo normal, transfusión de glóbulos cuando hubo caída de la hemoglobina, curas de lesiones y antibioticoterapia. El examen de proteína C del recién nacido resultó ser 2 %, y se obtuvieron niveles bajos de proteína C en el padre. Se confirma el diagnóstico de PF neonatal hereditaria.
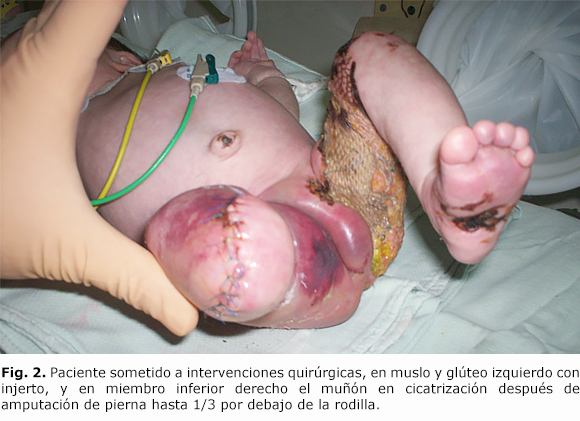
Algunas áreas afectadas tuvieron recuperación, pero otras fueron demarcando necrosis tisular con formación de gruesas costras (figura 1B). Se decidió el tratamiento quirúrgico con amputación del pie y pierna derecha hasta un tercio por debajo de la rodilla, por la especialidad de Ortopedia; y necrectomía y escarectomía con injerto en el muslo y glúteo izquierdo, por la especialidad de Cirugía Reconstructiva (figura 2) a los 21 y 24 días de edad respectivamente. La evolución de ambos procedimientos quirúrgicos resultó favorable, y los marcadores de infección evaluados por cultivos al ingreso fueron negativos.
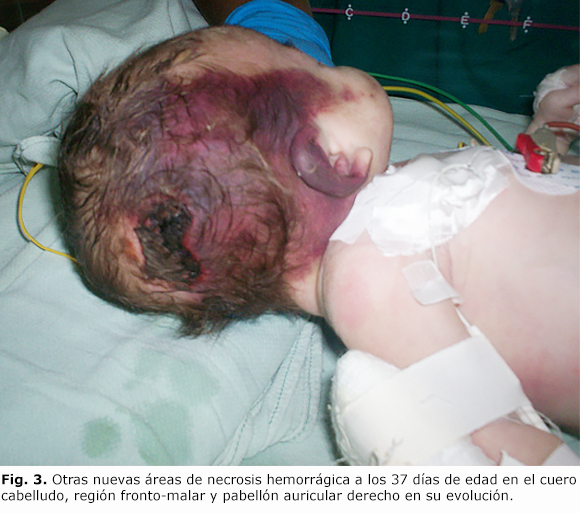
El paciente tuvo una evolución clínica favorable, con mejor aspecto general; sin embargo, a los 37 días de vida se produjeron otros eventos de trombosis en el cuero cabelludo, de un área ya afectada en región parieto-occipital, pero extendiéndose hacia hemicara derecha (figura 3). El coagulograma mostró nuevamente similares anormalidades. Su estado se fue deteriorando paulatinamente en los días subsiguientes, y fallece a los 41 días de edad. Los hallazgos necrópsicos muestran múltiples trombosis venosas y arteriales, y signos de infección sistémica.
DISCUSIÓN
El paciente presentado exhibe los elementos clínico-patológicos que caracterizan la PF, cuyo origen se debe a una deficiencia hereditaria de proteína C, lo cual condujo a la aparición de complicaciones trombóticas severas.
La PF fue descrita por primera vez en un neonato en 1962, y la causa se presumió ser un desorden hereditario, ya que 3 hermanos tuvieron similares lesiones,13 sin relacionarlo con la deficiencia de la proteína C, pero ilustra los hallazgos clínicos y alteraciones de exámenes de laboratorio característicos de este desorden, particularmente los referentes al coagulograma. Solo más tarde es que fue posible conectar los hallazgos clínicos de PF y la deficiencia de proteína C. El primer reporte de deficiencia de proteína C heredado en forma autosómica dominante fue descrito por Griffin en 1981,14 y 2 años más tarde, Branson reportó el primer caso de deficiencia severa de proteína C asociado a PF neonatal como manifestación clínica, y tratado exitosamente con reposición de proteína C.15
La PF tiene causas que por su origen se clasifican en congénitas o hereditarias, y adquiridas. Las causas adquiridas, por lo general, asociadas a infecciones, son provocadas por una diversidad de microorganismos que desencadenan una coagulopatía de consumo y deficiencia relativa de las proteínas C y S.2,3,5,16
Las causas congénitas o hereditarias se deben a deficiencias de las proteínas C y S homocigóticas, aunque pueden presentarse casos de carácter heterocigótico y co-hereditarias con otras trombofilias hereditarias.17,18 Hay 2 tipos de deficiencia de las proteína C: el más común es el tipo I, en el que los pacientes tienen tanto disminución de la proteína C, así como de su funcionalidad; y la tipo II, en la cual el paciente tiene una función alterada de la proteína C, pero su concentración plasmática es normal.17,19 En general, el déficit congénito de proteína C se describe como un rasgo autosómico dominante con penetrancia incompleta.1,6,17 Inicialmente considerada como una enfermedad monogenética, hoy día, basado en estudios de familias con trombofilia, que han mostrado una variabilidad manifiesta en la penetrancia del fenotipo, la trombofilia ha sido propuesta como una enfermedad oligogenética.
La deficiencia de la proteína C es causada por pérdida de la función de la mutación en el gen de la proteína C (PROC). Las mutaciones son muy heterogéneas, y la mayoría causadas por la sustitución de un nucleótido en la región del codón PROC.20 Se identificaron diversos factores de riesgo genético en pacientes con trombosis: la deficiencia de proteína C, de proteína S, el factor V de Leiden, la protrombina 20210G>A, así como los portadores de un grupo sanguíneo que no fuera O.1 Hay variantes de la enfermedad en las que, probablemente, solo los elementos biológicos no podrían justificar la inexplicabilidad de la herencia para el tromboembolismo venoso, como ocurre en este caso. La modificación de la cromatina, la regulación transcripcional y la eficiencia transnacional, pueden estar implicadas en la molécula de RNA; sin embargo, también se hace partícipe de esta variabilidad en la herencia a mecanismos epigenéticos. La hipótesis más reciente es que la presencia de variantes raras puede explicar la susceptibilidad genética del tromboembolismo venoso. El método más apropiado para determinarlas sería el estudio completo del genoma, lo que no es posible, pues habría que estudiar grandes poblaciones y sería extremadamente costoso.1
Este paciente comenzó con las manifestaciones de PF apenas con 48 h de vida, sin existir antecedentes de riesgo de infección prenatal y natal; tampoco se evidenció una clínica de sepsis, ni en los estudios microbiológicos se identificó algún microorganismo patógeno en distintas fuentes. El inicio temprano, junto con la determinación de la actividad de la proteína C en el neonato (2 %) y en uno de sus padres, sugieren el origen hereditario de su enfermedad.
La deficiencia de proteína C homocigota o heterocigota es muy rara, y causa enfermedad tromboembólica severa y PF en recién nacidos.21 La prevalencia de deficiencia de proteína C asintomática se reporta entre 1 en 200, a 1 en 500 individuos, la incidencia de deficiencia de proteína C clínicamente significativa, se ha estimado en 1 x 20 000 a 1 x 50 000;1,22 mientras que, la deficiencia de proteína C homocigótica, afecta aproximadamente 1 x 400 000, a 1 x 1 000 000 de nacidos vivos.18 En 22 años de trabajo (1992 hasta 2013) en el servicio de Neonatología de este hospital, se realizaron 10 131 ingresos, y este paciente ha sido el único en el que se ha diagnosticado PF por deficiencia hereditaria de proteína C.
Las proteínas C y S son glicoproteínas dependientes de vitamina K, con propiedades antitrombóticas, sintetizadas en el hígado. La proteína C es una proteasa sérica que circula como un cimógeno, después de la división proteolítica, por el complejo trombina/trombomodulina, sobre la superficie endotelial, la cual actúa inactivando los factores Va y VIIIa por proteolisis. La proteína C activada también tiene propiedades profibrinolíticas, por inhibición del plasminógeno activado. La proteína S actúa como un cofactor en este proceso, e inhibe así fenómenos trombóticos.6,22
El desarrollo de PF debido a deficiencia de proteína C, o proteína S homocigota, puede ser separada en 2 distintas fases. La primera fase es el período cuando las lesiones iniciales reversibles se desarrollan y crecen. Esta progresión reversible puede ser detenida y revertida con la administración de proteína C o proteína S. La segunda fase es un estadío irreversible, en el cual la lesión continúa para desarrollar una lesión necrótica, sea o no tratada con proteína C. Esta lesión irreversible finalmente desarrolla una extensa y gruesa lesión necrótica de la piel.23 Ese fue el curso que el paciente desarrolló por su enfermedad, con lesiones de trombosis, hemorragia y necrosis extensa.
En los aspectos terapéuticos de la PF, van der Horst desde 1962 aconseja el tratamiento con anticoagulantes.13 Branson y otros, en 1983,15 señalan como aspecto destacable que la enfermedad tenía contención con adecuada administración de plasma fresco, crioprecipitado y heparina; mientras que Estellés y otros, en 1984,24 muestran que aun un paciente homocigótico a la deficiencia de proteína C, puede sobrevivir con adecuada administración de plasma. Desde entonces la utilización de plasma fresco constituye un pilar fundamental del tratamiento de estos pacientes,6,25-27 lo cual incrementa los niveles de proteína C, además de tener este incremento un rol antiinflamatorio,28 junto con otros medicamentos anticoagulantes (heparina).6,29,30
Nuevos derivados de la sangre se han incorporado al arsenal terapéutico de la PF, tales como, concentrado altamente purificado de proteína C,1,6,8,27,31 proteína C reactivada recombinante8 y proteína C derivada del plasma no activada.12 El paciente motivo de este reporte recibió periódicamente plasma fresco y anticoagulantes (heparina), lo cual permitió, en algunos períodos de su estadía, llevar a la normalidad los valores del coagulograma. A pesar de ello ocurrieron fenómenos trombóticos severos que determinaron su evolución fatal.
En un estudio con la administración de proteína C derivada del plasma no activada (Ceprotin©), en niños mayores de 3 meses de edad, quienes sufrieron deficiencia de proteína C secundaria a infecciones u otros estados patológicos, se valoró la tasa de amputaciones digitales o de miembros con la administración del producto. Se encuentra que aunque disminuyeron las tasas de amputación, respecto a las reportadas en la literatura de los casos no tratados, fue inevitable ejecutar amputaciones en 16 % de estos niños.12 El tratamiento quirúrgico en este caso fue imprescindible realizarlo, debido a las extensas áreas de necrosis, fundamentalmente en glúteos y extremidades. Este tratamiento siguió los estándares recomendados,8 de un equipo multidisciplinario (neonatólogos, hematólogos, enfermeras, angiólogos, ortopédicos, cirujanos reconstructivo/caumólogos y anestesistas), centrando el manejo en evitar las infecciones asociadas a la atención hospitalaria y el soporte nutricional adecuado. Tal como está recomendado, el debridamiento, injerto y amputación que sufrió la paciente se realizó hasta que las áreas infartadas se demarcaron completamente de los tejidos circundantes, además de la administración de plasma fresco previo a las intervenciones, independientemente de las indicaciones periódicas que recibía de este derivado de la sangre. De hecho, la recuperación de las áreas injertadas y de la amputación de la extremidad fue favorable.
Se concluye que el paciente, por la precocidad de la aparición y las características clínicas típicas de la PF, en ausencia de sepsis o cualquier otro de los factores capaces de originar una deficiencia de proteína C, la determinación de la proteína C en el paciente y sus padres, su evolución posterior y los hallazgos necrópsicos, sustentan el diagnóstico de una deficiencia de proteína C hereditaria que ocasiona fenómenos tromboembólicos, responsables de las manifestaciones clínicas y curso posterior y fatal.
REFERENCIAS BIBLIOGRÁFICAS
1. Foy F, Moll S. Thrombophilia: 2009 Update. Curr Treat Options Cardiovr Med. 2009;11:114-28.
2. Fischer D, Allendorf A, Buxmann H, Weiss K, Schloesser RL. Purpura fulminans after therapeutic hypothermia in an asphyxiated neonate with Streptococcemia. Am J Perinatol. 2014;31(04):257-60.
3. Atay E, Akin M, Yüzkollar E, Atay Z, Dursun E, Ceran Ö. Neonatal purpura fulminans associated with early-onset gram-negative enterobacter septicemia: A case report. Int Pediatr. 2003;18(3):162-3.
4. Teertstra TK, Bergman KA, Benne CA, Albers MJ. Serratia marcescens septicemia presenting as purpura fulminans in a premature newborn. Eur J Clin Microbiol Infect Dis. 2005;24: 846-7.
5. Chalmers E, Cooper P, Forman K, Grimley C, Khair K, Minford A, et al. Purpura fulminans: recognition, diagnosis and management. Arch Dis Child. 2011;96(11):1066-71.
6. Price VE, Ledingham DL, Krümpel A, Chan AK. Diagnosis and management of neonatal purpura fulminans. Sem Fetal Neonatal Med. 2011;16:318-22.
7. del Giudice P, Chosidow O. Skin Manifestations of Systemic Bacterial Infections. In: Revuz J, eds. Life-Threatening Dermatoses and Emergencies in Dermatology. Berlin: Springer-Verlag Berlin Heidelberg; 2009. p. 123-31.
8. Faust SN, Nadel S. Purpura Fulminans. In: Revuz J, eds. Life-Threatening Dermatoses and Emergencies in Dermatology. Berlin: Springer-Verlag Berlin Heidelberg; 2009. p. 45-55.
9. Cassels-Brown A, Minford AMB, Chatfield SL, Bradbury JA. Ophthalmic manifestations of neonatal protein C deficiency. Br J Ophthalmol. 1994;78:486-7.
10. Khan NM, Al-Dohayan ND, Al-Batiniji FS. Bilateral leukocoria in a patient with homozygous protein C deficiency. Saudi Med J. 2007;28(7):1129-32.
11. van Schendel MP, Visser DH, Rammeloo LAJ, Hazekamp MG, Hruda J. Left pulmonary artery thrombosis in a neonate with left lung hypoplasia. Case Rep Pediatr [serie en Internet]. 2012 [citado 8 de febrero de 2015];article ID 314256. Disponible en: http://dx.doi.org/10.1155/2012/314256.
12. Piccini A, O’Marcaigh O, Mc Mahon C, Murphy C, Okafor I, Marcheselli L, et al. Non-activated plasma-derived PC improves amputation rate of children undergoing sepsis. Thrombosis Res. 2014;134:63-7.
13. van der Horst RL. Purpura fulminans in a newborn baby. Arch Dis Child. 1962;37:436-41.
14. Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, Wideman C. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68(5):1370-3.
15. Branson HE, Marble R, Katz J, Griffin JH. Inherited protein C deficiency and coumarin-responsive chronic relapsing purpura fulminans in a newborn infant. The Lancet. 1983;322(8360):1165-8.
16. Church J, Haram NH, Jones I, Hartnoll G. Purpura fulminans in an acute preterm neonate. Arch Dis Child Fetal Neonatal. 2012;98:F76-F77.
17. Khan S, Dickerman JD. Hereditary thrombophilia. Thrombosis Journal. 2006;4:15-31.
18. Yang JYK, Chan AKC. Pediatric Thrombophilia. Pediatr Clin N Am. 2013;60:1443-62.
19. Tridapalli E, Stella M, Capretti MG, Faldella G. Neonatal arterial iliac thrombosis in type-I protein C deficiency: a case report. Italian Journal of Pediatrics. 2010;36:23-5.
20. Bereczky Z, Kovacs KB, Muszbek L. Protein C and protein S deficiencies: similarities and differences between two brothers playing in the same game. Clin Chem Lab Med. 2010;48(suppl 1):S53-S66.
21. Marlar RA, Mastovich S. Hereditary protein C deficiency: a review of the genetics, clinical presentation, diagnosis and treatment. Blood Coagul Fibrinolysis. 1990;1(3):319-30.
22. Goldenberg NA, Manco-Johnson MJ. Protein C deficiency. Haemophilia. 2008;14(6):1214-21.
23. Marlar RA, Neumann A. Neonatal purpura fulminans due to homozygous protein C or protein S deficiencies. Semin Thromb Hemost. 1990;16(4):299-309.
24. Estellés A, García-Plaza I, Dasí A, Aznar J, Duart M, Sanz G, et al. Severe inherited “homozygous” protein C deficiency in a newborn infant. Thromb Haemost. 1984;52(1):53-6.
25. Sekiguchi K, Akiyoshi K, Okazaki N, Yamada H, Suzuki M, Maeda T, et al. PLEDs in an infant with congenital protein C deficiency: A case report (letter). Clin Neurophysiol. 2010;121:800-2.
26. Matsunaga Y, Ohga S, Kinjo T, Ochiai M, Ito N, Doi T, et al. Neonatal asphyxia and renal failure as the presentation of non-inherited protein C deficiency. J Perinatol. 2013;33:239-41.
27. Unal S, Gumruk F, Yigit S, Tuncer M, Tavil B, Cil O, et al. A novel mutation in protein C gene (PROC) causing severe phenotype in neonatal period. Pediatr Blood Cancer. 2014;61:763-4.
28. Esmon CT. Protein C anticoagulant system-anti-inflammatory effects. Semin Immunopathol. 2012;34:127-32.
29. Ficuccilli F, Lacobini M, Beglieri MR, Papoff P, Mancuso M, Caradonna A, et al. Purpura fulminans in the newborn. Report of two cases successfully treated with heparin and antithrombin III. Minerva Pediatr. 1997;49(12):571-7.
30. Hoffman R, Benz EJ, Silberstein LE, Heslop HE, Weitz JI, Anastasi J. Hematology: Basic Principles and Practice. Sixth Edition. Chapter 152. Disorders of Coagulation in the Neonate. New York: Churchill Livingstone Inc.; 2013. p. 2120-30.
31. Dreyfus M, Magny JF. Treatment of homozygous protein C deficiency and neonatal purpura fulminans with a purified. N Engl J Med. 1991;325(22):1565.
Recibido: 10 de junio de 2015.
Aprobado: 11 de agosto de 2015.
Bárbara Acosta Batista. Hospital Pediátrico “Juan Manuel Márquez”. Avenida 31 y 76, municipio Marianao. La Habana, Cuba. Correo electrónico: bacosta@infomed.sld.cu

