










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La malabsorción de glucosa-galactosa (MGG) es una enfermedad genética infrecuente. Es la consecuencia de una anomalía en el transportador intestinal específico. El cuadro clínico se caracteriza por una diarrea precoz que empieza en la edad neonatal y que puede poner en peligro el pronóstico vital del niño. El diagnóstico parece fácil cuando existen antecedentes familiares. 1
El objetivo de este artículo es describir una asociación infrecuente entre el síndrome de Down y la mala absorción de glucosa y de galactosa.
Presentación del caso
Paciente masculino de 3 ½ meses de edad con síndrome de Down, hijo de padres consanguíneos, sin antecedentes relevantes familiares o durante la gestación. Nacido a las 35 semanas de gestación, sin complicaciones, con 3 000 gramos de peso y alimentado con leche artificial. A partir del séptimo día de vida y durante una semana, presentó vómitos alimentarios abundantes y deposiciones diarreicas non stop explosivas sin sangre, con ruidos intestinales hiperactivos. El cuadro clínico empeoró con la utilización de las sales de rehidratación oral (SRO). En el décimo día, el niño fue hospitalizado por deshidratación severa y fue perfundido, lo que hizo que mejorara sensiblemente. Se sospechó alergia a la proteína de vaca (IgE: 0,12 > 0,1UI/ml), se prescribió una fórmula parcialmente hidrolizada sin proteínas de leche de vaca. El paciente fue rehospitalizado cuatro veces por el mismo motivo durante los siguientes tres meses y cada vez, el tránsito intestinal se mejoró simultáneamente con la eliminación de la alimentación oral y a la perfusión hidroelectrolítica. Se intentó varias preparaciones lácteas: (leche de cabra, leche a base de arroz, leche de soja, leche sin lactosa, leche hipoalergénica a base de aminoácidos libres) sin éxito. El niño se remitió a nuestro centro para valoración por gastroenterología pediátrica.
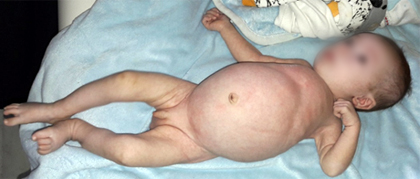
Los parámetros antropométricos demostraron una malnutrición severa tipo marasmo: peso 3 560 g (p3-p15), talla 51 cm (p3-p15), perímetro cefálico 38 cm (p3-p15), índice de masa corporal (IMC): 13,68 kg/m2 /p3. Se notó distención abdominal y miembros finos (Fig.1).
Se realizaron pruebas biológicas que dieron los siguientes resultados: hemoglobinemia: 9,9 g/dl, glucemia: 0,79 g/l, urea: 0,2 g/l, creatinina 6 mg/L, TGO: 22 UI/L, TGP: 23U/L, Na: 135 mmol/L, K: 4,7 mmol/L, albuminemia: 32 g/L, gases sanguíneos normales. Uroanálisis con densidad 1 025, pH 6 con parámetros negativos. Los estudios bacteriológicos y parasitológicos de las heces fueron normales y el pH fecal /5 (Fig. 2), el ionograma en materia fecal de 24 horas fue: sodio 0,5 mEq (1-10), potasio 2,6 mEq (8-22) y el cálculo realizado para distinguir entre diferentes causas de diarrea (Gap osmótico) dio aumentado: 168 mOsm/kg (50-125).

Dada la asociación de:
La consanguinidad.
Numerosos episodios de deshidratación con inicio temprano desde el periodo neonatal y de la malnutrición.
La diarrea osmótica con pH / 5, presencia de la glucosa, Gap osmótico elevado.
El fracaso de las preparaciones lácteas elementales y de las SRO.
Se consideró el diagnóstico de síndrome de malabsorción de glucosa y galactosa. Se instituyó una prueba terapéutica con fórmula a base de fructosa. Se logró la normalización del tránsito, se mejoró el estado nutricional con buena tolerancia a la fructosa lo que confirmó posteriormente el diagnóstico.
Discusión
La MGG congénita es una afección digestiva poco frecuente. Es un trastorno genético de herencia autosómica recesiva de carácter familiar como lo menciona Assiri.2 Los primeros autores que la describieron en el año 1962 fueron Laplaney Lindquist Meeuwisse,3 citados por Pascual Pérez y otros. Hasta hoy en día, se han publicado alrededor de 300 observaciones.1 La serie más larga citada en la literatura es de 33 pacientes.4 En cuanto a la población árabe, la cohorte más grande fue compuesta por 24 casos en Arabia Saudí. 5) En Argelia, ha sido recientemente publicada una serie de 4 casos.1
La glucosa y la galactosa son azucares básicos esenciales en el metabolismo celular y solicitan para su ingreso al enterocito, un transportador en la membrana celular, el sodio, dependiente de la glucosa transportador 1 (SGT1) de capacidad baja y afinidad alta. Esta proteína se ubica en el sistema gastrointestinal y es específica para la absorción de glucosa y galactosa en las células epiteliales del ribete en cepillo. Por otra parte, la mucosa yeyunal y la actividad de las oligosacaridasas en el borde en cepillo, son normales.6
Normalmente, se realiza un transporte acoplado, en el que entran simultáneamente al enterocito sodio y glucosa o galactosa. En las condiciones del caso presentado, el trasportador afectado es incapaz de efectuar esta acción en el íleon por sí solo. Este trastorno funcional se explica por numerosas mutaciones del cromosoma 22q12.3 en el gen SLC5A124,6,7 cuyas mutaciones descritas llegan a 56, entre ellas, dos nuevas: c.265G>A (p.G89R) y c.1304 G>A (p.G435D).8, 9)
Además, el transporte tubular de la glucosa se efectúa por medio del mismo transportador SGT1 y SGT2, cuyos defectos asocian a la MGG una glucosuria permanente o intermitente.10 Por otro lado, se reconocen dos tipos de transportadores: glucosa transportador 1 o GLUT2 y glucosa trasportador 5 GLUT5 que transportan tanto la glucosa como la fructosa. Esto se considera como la base de la dieta alternativa terapéutica en caso de MGG.
La MGG se manifiesta por diarrea grave non stop con heces acuosas, acidas (pH /5).4 La presencia y la acumulación de un soluto osmóticamente activo y no absorbido en el lumen producirá un aumento de la carga osmótica intraluminal, lo que induce diarrea osmótica con pH fecal bajo, es decir, ácido. Esta acidez es debida al hidrogeno y algas carbónicas liberados a partir de ácidos volátiles y ácido láctico tras la fermentación de los azucares acumulados por la flora intestinal.11 Es una diarrea de fermentación que aparece inmediatamente tras la ingesta de toda preparación que contenga lactosa, sacarosa o dextrina maltosa, lo que aumenta el Gap osmótico de la materia fecal.
La MGG puede revelarse por deshidratación aguda en periodo neonatal o por malnutrición más tarde, lo que puede retrasar el diagnóstico, como sucedió en nuestra observación.
Los diagnósticos diferenciales que pueden ser sospechados son esencialmente los de: la diarrea neonatal severa como clorhidrorrea congénita, intolerancia a la lactosa, alergia a la leche de vaca y diarrea infecciosa.12,13
Las manipulaciones dietéticas constituyen la piedra angular del diagnóstico, para confirmarlo también se puede recurrir a las pruebas de absorción oral con determinación del hidrogeno espirado tras la ingesta de glucosa o galactosa, aunque son pruebas difíciles de realizar en el niño,1 no obstante, la existencia de un caso similar intrafamiliar lo facilita más. Esta afección puede acompañarse de trastornos metabólicos e hidroeléctricos, si bien son poco frecuentes o extremadamente raros. Así nombramos hipernatremia con o sin gangrena, hipercalcemia con nefrocalcinosis, entre otros.1,14
La particularidad de nuestro caso es la asociación de la MGG con el síndrome de Down, que según nuestro conocimiento, es la primera vez que tal relación es descrita. Hubiera sido útil completarla con un estudio genético en búsqueda del tipo de mutación.
La única terapéutica consiste en una dieta preparada solo con fructosa, que favorece la desaparición total de la diarrea así como un posterior crecimiento normal. Se recurre a una preparación láctea especialmente adaptada: la galactomina 19 azucarada exclusivamente con fructosa. También, se deben prohibir medicamentos como las SRO, los antibióticos (la amoxicilina) por su contenido en glucosa.
Globalmente, la evolución es favorable con la adquisición de tolerancia a la leche, al azúcar o al almidón. Esto puede explicarse, por un lado, por la adaptación de la flora intestinal y por otro, a la persistencia de una actividad del transportador, aunque sea residual.14
Podemos concluir que es importante tener en cuenta la malabsorción de glucosa y de galactosa dentro de los diagnósticos diferenciales de las diarreas acuosas congénitas. El diagnóstico precoz y la dieta adecuada con fructosa evitan deshidratación y malnutrición. La particularidad de nuestro caso es la asociación de la malabsorción de glucosa y de galactosa con el síndrome de Down, que, según nuestro conocimiento, es la primera vez que se describe y podría aumentar la morbilidad.

