










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La miocardiopatía hipertrófica (MCH) es un trastorno hereditario caracterizado por hipertrofia ventricular, no explicada por otra lesión orgánica del corazón o por trastorno funcional con hipertrofia reactiva a sobrecarga sistólica por lesión valvular o hipertensión arterial.
La primera referencia que resume una serie de 8 pacientes entre 14 y 44 años de edad fue publicada por Donal Theare en 1957; en ella estableció la relación entre los hallazgos anatomopatológicos, la clínica y la electrocardiografía.1) Hasta hoy, con el desarrollo de la ecocardiografía en la década de los años 70, siglo xx, y el vertiginoso desarrollo de la genética desde los años 90, se ha podido establecer hasta más de 1400 mutaciones en más de 10 genes que explican los hallazgos clínicos y ecocardiográficos de estos pacientes.1,2
Las mutaciones ocurren en genes de las proteínas de la sarcómera en 40 a 60 % de los pacientes, con trasmisión hereditaria, autosómica dominante; hasta 10 % por enfermedades por errores del metabolismo, enfermedades mitocondriales, neuromusculares y síndromes genéticos. Los pacientes con antecedentes familiares presentan mutación de la cadena pesada de beta miosina (MYH7), 35-50 %; de la proteína C ligada a la miosina (MYBPC3) de 15 a 25 % o de la troponina cardiaca tipo 2(TNNT2) en 15-20 %.3) En 30 % de los pacientes no es identificable la etiología. Su origen podría encontrarse en mecanismos epigenéticos.4,5
Las fibras miocárdicas se disponen de manera anormal y como consecuencia, se expresan trastornos intrínsecos de los mecanismos de la velocidad y la fuerza de la contracción y de trastornos de la relajación y de la velocidad de relajación por fibrosis intersticial, además de alteraciones de la vascularización coronaria.(6 7)
La prevalencia de la MCH es de 0,2 % o de 1/500 aunque se presume mayor por la presencia de lesión genética sin expresión fenotípica.2
El debut clínico puede ser por arritmia cardiaca, por manifestaciones obstructivas del tracto de salida del ventrículo izquierdo (TSVI), por lesiones isquémicas o por disminución de la capacidad funcional con hipotensión arterial y disminución de la frecuencia cardiaca al ejercicio por desregulación autonómica.8 La MCH es la principal causa de muerte súbita en los jóvenes.9) En Cuba aparece como causa de defunción en ascenso según avanzan las décadas de vida.10
El diagnóstico se basa en los antecedentes familiares, los síntomas o signos de lesión cardiaca y los hallazgos ecocardiográficos. La ecocardiografía determina el diagnóstico por la medición de la pared posterior del ventrículo izquierdo (VI) y la hipertrófia del tabique interventricular con hipertrofia mayor a la segunda desviación estándar para las dimensiones correspondientes a la superficie corporal y en algunos casos, el movimiento anormal de la valva anterior de la mitral hacia el TSVI. La distribucón simétrica o regular o la disposición asimétrica o segmentaria de esta hipertrofia se puede diagnosticar además, con el uso de la angiotomografía o la resonancia magnética nuclear.6
Se define como obstructiva o no obstructiva si interfiere o no, con la salida del flujo sanguíneo a través de la salida del ventrículo izquierdo. Existe obstrucción cuando el gradiente máximo en el TSVI excede los 30 mm Hg en reposo y 50 mm Hg al ejercicio y se relaciona con complicaciones evolutivas.11) A la obstrucción puede contribuir un movimiento de la válvula anterior de la mitral (SAM, por sus siglas en inglés) durante la sístole hacia el TSVI provocado por una suerte de succión o arrastre conocido como efecto “Venturi”, lo que provoca un apoyo de la valva sobre el tabique e insuficiencia mitral; puede coexistir una inserción anómala del músculo papilar anterior o su hipertrofia mismo.8
La detección de esta lesión previa al desarrollo de los síntomas y su tratamiento pudiera mejorar la respuesta terapéutica y disminuir el ritmo de hipertrofia y con ello, mejorar la expectativa de vida de estos pacientes.
Existen muy pocas referencias nacionales del estudio de esta entidad.12,13 El Cardiocentro Pediátrico “William Soler” (CPWS) es el centro de referencia nacional para el tratamiento de las cardiopatías congénitas y en los últimos diez años se han ingresado pacientes con este diagnóstico. Este trabajo se propone mostrar la prevalencia de la miocardiopatía hipertrófica en la práctica del Cardiocentro Pediátrico “William Soler” y sus formas de presentación.
Métodos
Se realizó un estudio longitudinal, retrospectivo, observacional desde enero de 2010 hasta 2020.
Se revisaron las bases de datos de los registros médicos del CPWS y se seleccionaron todos los pacientes ingresados con el diagnóstico de miocardiopatía hipertrófica. Se excluyeron aquellos a quienes se les detectó una causa primaria de la hipertrofia como estenosis aórtica, hipertensión arterial o estenosis subaórtica discreta.
Se recogieron variables demográficas: sexo y edad del debut clínico de la enfermedad, edad al final del estudio y tiempo de seguimiento. Variables clínicas: síntoma principal, y ecocardiográficas: obstrucción al TSVI, grosor de la PPVI y del TIV y la presencia de SAM, en cada uno de los ingresos en cada paciente. Se dividieron para su análisis en dos grupos, los que presentaban MCH obstructiva (MCHO) y los que presentaban MCH no obstructiva. Los rangos de edades se corresponden con la pirámide poblacional.14
Se construyeron distribuciones de frecuencias y tablas de contingencia. Las variables cualitativas se resumieron en porcentajes y las cuantitativas en medias y desviaciones estándar. Se analizaron con la prueba de Ji-cuadrada(χ²) con corrección de Yates si fue necesario y la prueba t de Student previo análisis de homogeneidad de varianzas, respectivamente. Se utilizó el programa estadístico XLSTAT para Excel de Addinsoft versión 2019. Todos los contrastes estadísticos se asumieron con una confianza de 95 %.
Se contó con la aprobación del consejo científico de la institución y de su comité de ética. Se respetaron los principios básicos de la Declaración de Helsinki para la investigación biomédica.
Resultados
Se detectaron 86 ingresos con el diagnóstico de MCH de los 29 304 ingresos de la institución en diez años de trabajo, lo que constituye 0,29 %.
Estos ingresos correspondieron a 21 pacientes, de los que 12 presentaban una MCHO y 9, MCH no obstructiva. En el grupo obstructivo 50 %, y en el no obstructivo 44 % fue del sexo masculino.
Tres (33 %) de los nueve pacientes con lesiones no obstructivas presentaron características asimétricas. De las obstructivas, 8 (66 %) eran asimétricas.
Del grupo de pacientes con obstrucción, 42 % (5 pacientes) se diagnosticaron con menos de 3 años de edad. No hubo diagnóstico en menor de 3 años de edad en los que no tenían componente obstructivo (Fig.1).
A los 18 años de edad, se diagnosticó el paciente mayor de esta investigación y se siguió por un año hasta el final del estudio. La paciente mayor al final del estudio tenía 27 años. El tiempo promedio de evolución fue de 12,8 años (1,4-26,1).
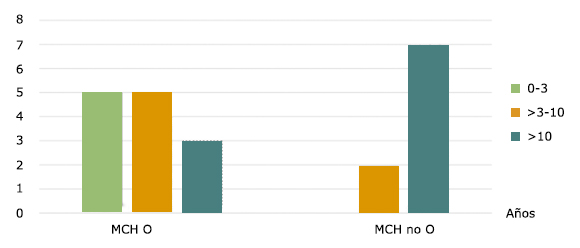
Se encontraron antecedentes genéticos hereditarios en tres de los 21 pacientes (14,3 %), dos varones con MCHO; uno de 9 años con antecedentes de una hermana y un tío con MCH; el otro de 10 años cuyo padre también tenía diagnóstico de MCH y una paciente femenina de 7 años de edad con MCH no obstructiva, cuyo abuelo paterno sufrió la enfermedad.
En cuatro pacientes hubo asociación con síndrome genético, dos de ellos con síndrome de Costello, una paciente que fue diagnosticada a los 3 meses de edad, con lesión obstructiva que requirió tratamiento quirúrgico y un paciente masculino diagnosticado a los 3 años de edad con obstrucción y tratamiento médico. Los otros síndromes asociados fueron en dos pacientes femeninas que presentaban MCHO, una con síndrome de Down y otra con síndrome de Ehlers-Danlos.
Once pacientes (52 %), fueron asintomáticos en el momento del diagnóstico, de ellos 8 con características obstructivas (66,6 %). El diagnóstico se realizó por sospecha al examen físico (Tabla 1).
Tabla 1 Relación síntomas-obstrucción
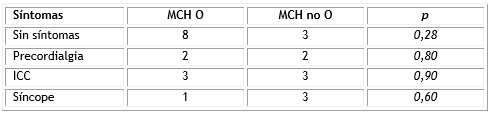
MCH O: miocardiopatía hipertrófica obstructiva; MCH no O: miocardiopatía hipertrófica no obstructiva; ICC: insuficiencia cardiaca.
Se detectaron diferencias significativas entre ambos grupos, en el gradiente del TSVI y en la edad de la detección de la lesión. No se encontraron diferencias en las medidas del tabique interventricular (TIV), de la pared posterior del VI (PPVI) o en la presencia de movimiento anormal de la valva anterior de la mitral (SAM) (Tabla 2).
Tabla 2 Resumen de las variables analizadas
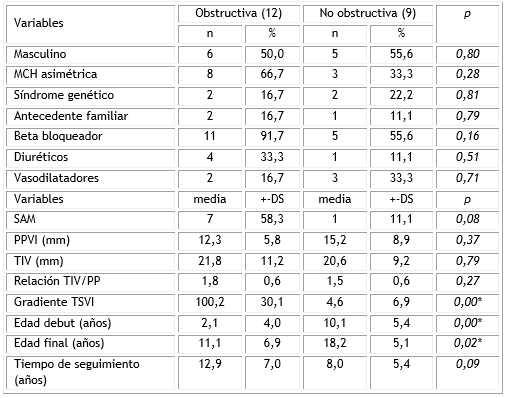
MCH: miocardiopatía hipertrófica; SAM: movimiento anterior de la mitral, PPVI: pared posterior del ventrículo izquierdo; TIV: tabique interventricular; TSVI: tracto de salida del ventrículo izquierdo, DS: desviación estándar,
*p<0,05
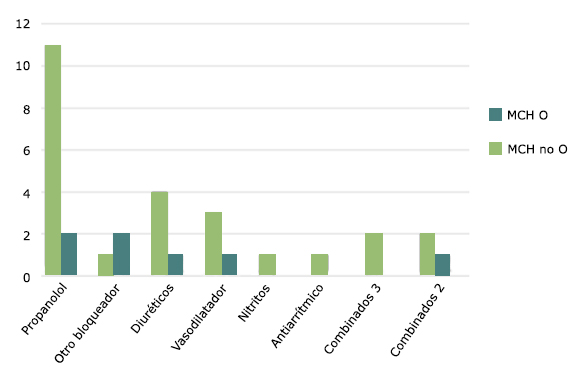
El tratamiento médico más utilizado fue el propranolol. Un paciente se trató con atenolol. Los diuréticos, vasodilatadores y antiarrítmicos se utilizaron con menor frecuencia (Fig. 2).
Los gradientes en TSVI disminuyeron en todos los pacientes con obstrucción con tratamiento médico o con tratamiento quirúrgico en 4 pacientes. Fue significativa la diferencia con el grupo que recibió cirugía para la liberación de dicha obstrucción (Tabla 3).
Tabla 3 Variación de los gradientes en las miocardiopatías obstructivas
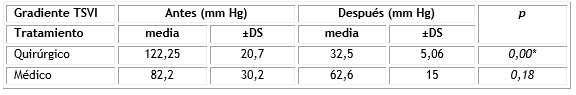
TSVI: tracto de salida del ventrículo izquierdo; *p<0,05.
Dos de pacientes, presentaron bloqueo aurículo ventricular posoperatorio que obligó a la colocación de marcapasos. A dos pacientes con MCH no obstructiva, que debutaron con síncope y taquicardia ventricular, se les colocó desfibrilador automático (DAI), ambos con lesiones no obstructivas, un varón de 16 años con MCH asimétrica y una paciente de 17 años con MCH simétrica.
No hubo fallecido en la serie estudiada en un tiempo entre 8 y 12 años de evolución (Tabla 2).
Discusión
Se estima que la prevalencia de la MCH en la población adulta en general es de 0,16 % (1: 625 individuos) a 0,29 % (1: 344).8
El número de pacientes diagnosticados en un centro de referencia nacional pudiera acercarse a la prevalencia de esta enfermedad, aunque se trata de una población pediátrica, en este trabajo fue mayor al encontrado por Márquez en un estudio de 15 años en el Instituto Nacional de Cardiología Ignacio Chávez de Ciudad de Méjico15 y mucho menor a la publicada en un estudio multicéntrico italiano de cuatro décadas que informó 6,1 %.16
En más de 5 millones de pacientes, se informa 0,07 %; con aumento de la prevalencia en la medida que aumentaron las décadas de vida de los sujetos estudiados.17) La prevalencia de la MCH en el período de 5 años fue de 2,35 %, en otro informe en el que la media de edad fue de 54 años.18
No hubo predominio de sexo en el estudio del CPWS. Se afirma que no existen diferencias en las primeras décadas de la vida y se refiere un aumento del predominio masculino luego de los 30 años de edad.17) Se registra una relación masculino femenino de hasta 5,5:1 y se plantea congruencia esto con la mortalidad.19,20
En el Cardiocentro Pediátrico predominó la MCH obstructiva. A diferencia de otros, (19 que tienen predominio del diagnóstico no obstructivo en 66 % de 59 pacientes.
Predominaron las lesiones asimétricas (52,3 %), en coincidencia con otros autores 16,21) que también encontraron un predominio de lesiones asimétricas aunque en porcentajes mayores, de 93 y 85 %.
En el presente estudio, la edad media de diagnóstico fue de 6,1 (± 4,7) años, menor que en otras publicaciones en que se plantea alrededor de los 12 (7-14) años de edad;16 coincidiendo con 20 % de los artículos seleccionados en un metaanálisis.22 Se plantea además, que los pacientes de menor edad presentan características obstructivas, coincidente con este reporte Cardiocentro Pediátrico y se asume como factor de riesgo para eventos cardiovasculares agudos o arritmias graves en su evolución a mediano y largo plazo.16
El bajo porcentaje de antecedentes familiares con afección por MCH de esta serie contrasta con otra, que informa hasta 83 % con familiares de primer orden y 10 % de segundo orden.21) El diagnóstico con determinación genética y el pesquisaje familiar pudiera incrementar el diagnóstico genotípico sin expresión fenotípica familiar.
La asociación de MCH en dos pacientes con síndrome de Costello se corresponde con la incidencia rara de este síndrome incluido en las llamadas rasopatías en los que se incluyen los síndromes de Noonan y Leopard en quienes hasta 71 % presenta MCH.23,24,25
No se registró en este estudio algún episodio de muerte súbita recuperado, aunque sí se reportó el síncope. En estudios de metanálisis se refiere como factor de riesgo para este evento el antecedente de síncope o de obstrucción severa al tracto de salida del VI. 16,22,26
Más de la mitad (57 %) de los pacientes aquí estudiados eran asintomáticos en el momento del diagnóstico, el cual se realizó por sospecha en el examen físico. Otros estudios también notifican asintomáticos hasta en 68 % de sus pacientes en el momento del diagnóstico16 y asintomáticos o con síntomas muy ligeros hasta 95 % de los pacientes estudiados.27)
La prevalencia basada en estudios de pesquisa ecocardiográfica es mayor que la prevalencia clínica.17,21 La sospecha clínica y el pesquisaje, pudieran ayudar a un diagnóstico temprano, tratamiento y mejor evolución clínica. A igual que en este informe, el síncope se presenta con menor frecuencia en otras series 16,19
Los bloqueadores de los receptores beta adrenérgicos, se han usado desde 1960 para controlar los síntomas en los pacientes con MCH. El más utilizado es el propranolol, sobre la base de la experiencia del CPWS. Su efectividad se ha demostrado en la disminución del dolor anginoso y la disminución de síntomas respiratorios durante el esfuerzo atribuibles al fallo ventricular izquierdo. Los efectos inotrópico y cronotrópico negativos producen disminución del consumo de oxígeno miocárdico y además mejoran la diástole mediante su prolongación, más que por una acción lusitrópica directa. 4,8,11,26,27
Las recomendaciones actuales para pacientes pediátricos no incluyen antagonistas del calcio o nitratos, que sí se recomiendan en el adulto. El verampamilo se recomienda con cautela en los niños con lesión no obstructiva para reducir las presiones interventriculares, disminuir la frecuencia cardiaca y con eso aumenta el periodo de llenado ventricular,4,11pero no existe esta experiencia en la serie revisada.
El uso de vasodilatadores es controversial. Mejoran la perfusión sistémica y disminuyen la insuficiencia mitral concomitante aunque incrementan el gradiente en el TSVI y disminuyen la presión diastólica y la perfusión coronaria, lo que favorece la isquemia del miocardio hipertrófico,11) sin embargo, fue utilizado, con buenos resultados, como único tratamiento o combinado con propranolol en la serie que se describe del CPWS. También se encontraron pacientes con lesión obstructiva e indicación de diuréticos, lo cual no se recomienda si no para lesiones no obstructivas y con limitación de la capacidad funcional.4
La amiodarona, el sotalol, además del beta bloqueador pueden ser utilizados para los episodios de arritmia,11) tal y como se usó en uno de los pacientes referidos en este informe.
Los síntomas de la obstrucción del TSVI mejoran o desaparecen en 90 % de los pacientes luego de la cirugía.11) Los resultados del Cardiocentro Pediátrico muestran una reducción significativa del gradiente luego de la cirugía, de 122 ± 20 mm Hg a 32 ± 5 mm Hg; mayor al resultado de un estudio que recoge la reducción del gradiente de 95±36 mm Hg a 12±6 mm Hg (p= 0.001). (11 ) La diferencia pudiera estar justificada por un protocolo que indica la intervención más temprano.
Castedo plantea que 5 % de los pacientes con MCHO no responden a tratamiento médico y tienen indicación quirúrgica.28) En el grupo del CPWS, se operó 33 % de los pacientes con MCHO basados en la existencia de gradiente de riesgo con o sin respuesta al tratamiento médico. La técnica quirúrgica consiste en una miectomia transaórtica del TIV acompañado o no de plastia de la valva anterior de la mitral.29
La reducción del gradiente a la salida del VI a través de la cirugía, ha demostrado una reducción de la mortalidad a largo plazo comparable a la de pacientes que no han necesitado de cirugía.3,27,30,31,32
El comienzo de la enfermedad en la edad pediátrica no se relaciona con la arritmia, la cual aparece en la adolescencia. Es por esto que la colocación de DAI no ofrece protección en edades tempranas.16) El riesgo de episodios de fibrilación ventricular y muerte súbita aumentan con la historia de sincope y de hipertrofia severa, que aumentan con la edad.33 Esta constituyó la indicación en dos adolescentes de esta serie, en coincidencia con laspublicadas,4,7,16,33) además de la historia familiar de muerte súbita.33
La indicación en el Cardiocentro Pediátrico de inserción de marcapasos fue por complicación posoperatoria, no se indicó como variante de tratamiento, relacionada con el tiempo de evolución y daño miocárdico como en otros estudios que incluyen adultos con MCH.4,11,34
El diagnóstico antes del año de edad es un factor de riesgo de mortalidad. 25 En este estudio, aunque hubo lactantes diagnosticados de naturaleza obstructiva, no hubo fallecido en el tiempo de seguimiento.
El carácter retrospectivo limita este estudio y permite recomendar, realizar análisis prospectivos para la evaluación de las estrategias terapéuticas e introducir en la práctica de la red cardiopediátrica la pesquisa de la miocardiopatía hipertrófica en la atención primaria de salud.
La miocardiopatía hipertrófica es un motivo de ingreso inusual en la práctica cardiopediátrica. La sospecha clínica y la pesquisa hacen posible la detección temprana en pacientes asintomáticos. El diagnóstico y seguimiento es posible por ecocardiografía y las opciones de tratamiento aplicadas en el Cardiocentro Pediátrico “William Soler”, coinciden con los consensos internacionales y han permitido la disminución del riesgo de muerte súbita.
Se concluye que la miocardiopatía hipertrófica tiene una baja prevalencia en la práctica cardiopediátrica. Los síntomas tempranos se corresponden con la variedad obstructiva. Su diagnóstico temprano y el tratamiento específico, permite garantizar mejor calidad y expectativa de vida a los portadores de esta afección.

