










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Las genodermatosis constituyen un grupo de afecciones clínicas muy heterogéneas, cuyas principales manifestaciones radican en la piel y sus anejos. Presentan como elemento común su condicionamiento genético.1) Se consideran enfermedades raras por su baja prevalencia, afectan menos de cinco personas por 10 000 habitantes.2)
Las ictiosis (Orpha 79354)3) hereditarias componen un grupo heterogéneo de afecciones caracterizadas por la presencia de escamas e hiperqueratosis en todo el cuerpo, a menudo asociadas con la inflamación de la piel.4
La ictiosis vulgar constituye una genodermatosis con patrón de herencia autosómica dominante (AD), puede ser congénita o comenzar en los primeros meses de vida con escamas finas y adherentes que respetan los pliegues de flexión.5) Se considera la más frecuente de todas las ictiosis y se observa en un caso por 300 personas.6) La enfermedad se produce por una mutación con pérdida de la función del gen filagrina en el locus 1q21.3, que resulta en una disfunción de la barrera epidérmica.7
El síndrome de Ehlers Danlos (Omim 130000, Orpha 98249)8,9) constituye un grupo heterogéneo de genodermatosis que afectan el tejido conectivo, se caracteriza por hipermovilidad articular, hiperextensibilidad de la piel y fragilidad tisular.10) La causa de este síndrome radica en las mutaciones de los genes que codifican el colágeno fibrilar tipo I, III y V, o en las enzimas comprometidas con la modificación postranslacional de dichos colágenos. La forma clásica resulta la más frecuente con una incidencia estimada de 1: 5000 nacimientos.11
Estos dos variados grupos de genodermatosis no comparten manifestaciones cutáneas ni extracutáneas similares. Es infrecuente encontrar concomitancia de ambas dermatosis en un mismo paciente, y la heterogeneidad clínica implica complejidad en el diagnóstico.
El objetivo de este trabajo fue exponer un caso que presentó ictiosis vulgar asociada con el síndrome de Ehlers Danlos tipo clásico, en el que el análisis del árbol genealógico contribuyó a orientar el diagnóstico.
Presentación del caso
Paciente femenina, de 10 años de edad, de procedencia urbana, con antecedentes de asma bronquial. Desde los primeros años de la infancia (3 a 4 años) su madre notaba que la piel de las extremidades, fundamentalmente en las piernas, se tornaba seca y escamosa, y empeoraba en los meses de invierno. Además de este cuadro, a la edad de nueve años presentó una subluxación espontánea de la escápula humeral derecha y comenzó a atenderse con la especialidad de ortopedia. Llamó la atención en ese momento que, además, presentaba hiperextensibilidad de la piel, y se remitió, por ello, a la consulta especializada multidisciplinaria de genodermatosis (especialidades de dermatología, genética médica clínica, ortopedia, hematología y psicología).
Este trabajo se sometió a la aprobación del Comité de Ética de la Investigación y el Consejo Científico del Hospital Pediátrico Provincial Docente “Mártires de Las Tunas” como institución ejecutora, acorde con los principios de la Declaración de Helsinki. Para la participación en el estudio y la obtención de las fotos, por los autores, se consiguió el consentimiento informado por escrito de los padres de la paciente.
La paciente presentaba:
Antecedentes patológicos personales: asma bronquial.
Antecedentes patológicos familiares: piel ictiosiforme (madre, tía y abuela maternos) con hiperextensión articular (padre, tía, tío, primo y abuela paternos)
Examen dermatológico: cuadro cutáneo monomorfo, localizado en cara anterior de las piernas, dado por lesiones escamosas ictiosiformes; presenta, además, aumento de las líneas palmares (fig. 1Ay B) .
Muestra hiperextensibilidad de la piel presente en orejas de discreta distención, pliegue del cuello, de la piel del tronco abdominal y de la piel del codo, mayores de 3 cm (fig. 2A) .
Examen del sistema osteomioarticular: hipermovilidad articular facilitada por hiperextensión de los miembros superiores mayor de 180 grados, aposición del pulgar (fig. 2B y C) , protuberancia del hemitorax izquierdo, presencia de genus valgus y pies planos.
Examen oftalmológico: presencia de astigmatismo, no ectopia del cristalino.
Fondo de ojo: papilas bien definidas, red vascular normal, mácula con brillo foveal, no opacidades del cristalino, no malformaciones oculares.
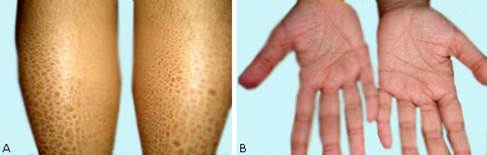
Fig. 1 - Lesiones de ictiosis vulgar. A: escamas ictiósicas localizadas en cara anterior de las piernas. B: aumento de los pliegues palmares.
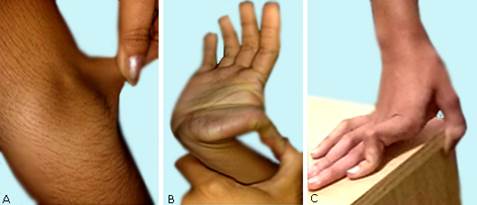
Fig. 2 - Lesiones de síndrome de Ehlers Danlos. A: hiperextensibilidad de la piel. B y C: hipermovilidad articular.
Estudios hematológicos: dentro de los parámetros normales con: hemoglobina: 133 g/L; leucos: 7,8 x 109/ L con diferencial polimorfos: 0,53; linfocitos:0,32; eosinóficos: 0,00; plaquetas: 250 x 109/L, coagulo retráctil, plaquetas agregadas; eritrosedimentación: 30 mm/ H.
Electrocardiografía y ecocardiografía: sin alteraciones.
Estudio histopatológico de lesiones ictiósicas de las piernas: hiperqueratosis ortoqueratósica compacta, tapones córneos, capa granular reducida e infiltrado linfocitario perivascular en la dermis. Sugestivo de ictiosis vulgar.
En el árbol genealógico, teniendo en cuenta los antecedentes familiares descritos, se observó la presencia de ictiosis vulgar que afectó a varios miembros en la familia materna y el síndrome de Ehlers Danlos tipo clásico, que afectó a varios miembros en la familia paterna (fig. 3).
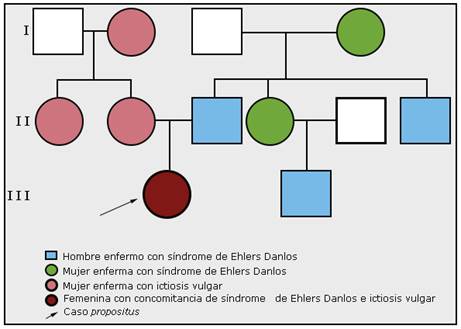
Fig. 3 - Árbol genealógico, donde se constata en varias generaciones, la presencia de ictiosis vulgar en miembros en la familia materna, y síndrome de Ehlers Danlos en miembros de la familia paterna.
Conducta terapéutica
Educación a la paciente y la familia en relación con el uso de emolientes para evitar puertas de entrada que acarreen infecciones de la piel y evitar traumatismos innecesarios en la piel debido a los riesgos de una mala cicatrización.
Terapia de rehabilitación para la tonificación de la musculatura y educación a la paciente en relación con los ejercicios físicos requeridos para evitar las luxaciones.
Tratamiento tópico de las lesiones ictiósicas. Uso de predneomin ungüento, dos veces al día.
Vitaminoterapia: multivitaminas 1 tableta/día oral, ácido fólico 1 mg/día oral y vitamina C 500 mg/día oral.
Seguimiento en consulta multidisciplinaria de atención a pacientes con genodermatosis.
Discusión
Se trata de una paciente que comenzó con lesiones de piel ictiosiformes desde edades tempranas, además de hiperextensibilidad de la piel e hipermovilidad articular que le provocaba luxaciones espontáneas. Presentaba antecedentes familiares maternos de piel ictiosiforme y antecedentes paternos de hipermovilidad articular.
Las ictiosis, conocida como piel de pescado, constituyen un grupo de enfermedades en la que los mecanismos hemostáticos en el que el quinecismo y la diferenciación de las células epidérmicas se presenta alterado, y aparecen escamas que semejan las escamas de pescado. El desorden que afecta la cornificación conlleva a la aparición de prurito, y un incremento de las infecciones epidérmicas; los síntomas y signos suelen empeorar durante el invierno.1
En 2009 un grupo de expertos desarrolló una nueva clasificación de consenso basada principalmente en las características clínicas, en los aspectos fisiopatológicos y moleculares descubiertos hasta el momento. Esta clasificación identificó 36 tipos de ictiosis, las cuales se dividieron en subgrupos de acuerdo con la presencia o no de compromiso extracutáneo, frecuencia de la enfermedad y patrón de herencia. Las no sindrómicas (que cursan solo con manifestaciones cutáneas) y las sindrómicas (que se presentan con manifestaciones en otros órganos).12
Las ictiosis no sindrómicas no manifiestan rasgos extracutáneos, salvo retraso del crecimiento. Estas son:
Ictiosis vulgar: con herencia AD, aparece generalmente después de los 2 a 6 meses, con descamación que afecta fundamentalmente las extremidades, existe aumento de las líneas palmoplantares.
Ictiosis recesiva ligada al cromosoma X (IRLX): al nacimiento puede presentar descamación leve o eritrodermia o membrana colodión leve. En la infancia o edad adulta aparece descamación generalizada de escamas finas o romboidales de color marrones o grises que no afectan los pliegues.
Ictiosis laminar: de herencia autosómica recesiva (AR): el niño nace envuelto en una membrana colodión, presenta ectropión y eclabio. Meses más tarde aparecen placas queratodérmicas generalizadas de gran tamaño, con queratodermia palmoplantar (QPP), hipohidrosis. Puede producir alopecia cicatrizal.
Eritrodermia ictiosiforme congénita: con herencia AR, desde el nacimiento presenta eritrodermia leve con descamación fina, hipohidrosis y puede producir alopecia cicatrizal.
Ictiosis en arlequín: con herencia AR. el niño nace envuelto en una membrana colodión en forma de placas que semejan una armadura, se acompaña de ectropión, eclabio, contracturas; le sigue una eritrodermia con descamación fina, alopecia cicatrizal, hipohidrosis y son frecuentes las infecciones bacterianas. Es una forma grave que puede ser letal.
Ictiosis epidermolítica: con herencia AD, comienza al nacimiento con eritrodermia, ampollas o erosiones, descamación leve, luego le sigue la formación de escamas gruesas y verrugosas acentuadas en áreas de fricción, puede presentar QPP y colonización bacteriana frecuente.
Ictiosis epidermolítica superficial: con herencia AD, comienza al nacimiento con eritrodermia, erosiones o ampollas superficiales diseminadas.
Ictiosis en confeti: con herencia AD, presenta desde el nacimiento eritrodermia con descamación fina, QPP y posteriormente, aparecen lesiones puntiformes similares al confeti.
Queratodermia por alteración de la loricrina: con herencia AD, al nacimiento el cuadro resulta similiar a la eritrodermia ictiosiforme congénita, luego aparece QPP en panal de miel con constricción digital (seudodactilólisis espontánea) y descamación generalizada leve que se acentúa en las articulaciones.
Eritroqueratodermia variable: con herencia AD, pueden aparecer signos cutáneos persistentes al nacimiento o en el primer año dados por parches eritematosos transitorios migratorios con descamación fina y placas hiperqueratóticas fijas y bien delimitadas, con o sin QPP.
Enfermedad de descamación cutánea: con herencia AR, al nacimiento el cuadro es similar a la eritrodermia ictiosiforme congénita, luego queda una eritrodermia generalizada con descamación exfoliativa migratoria y son frecuentes las infecciones cutáneas.
Queratosis lineal-ictiosis congénita-queratodermia: con herencia AR, al nacimiento se presenta una membrana colodión leve y luego se producen placas y pápulas hiperqueratóticas en los pliegues cutáneos, QPP con bandas de constricción digital.13
Los síndromes ictiosiformes presentan lesiones de tipo ictiodiforme que pueden ser de tipo eritrodérmico asociado a otras manifestaciones extracutáneas; algunos de estos son:
Síndrome de Netherton: presenta herencia AR, caracterizada por ictiosis lineal circunfleja con placas de doble borde, tricorrexis, diátesis atópica con aumento de la IgE, eosinofília y retraso del crecimiento.
Tricotidistrofia: presenta herencia AR, con descamación fina, tricosquisis o tricorrexis, retraso del desarrollo, cataratas, dismorfismo facial, anomalías óseas y gonadales; resultan frecuentes las infecciones recurrentes.
Ictiosis folicular-atriquia-fotofobia: con herencia recesiva ligada al cromosoma X, el paciente nace envuelto en una membrana colodión, luego se produce hiperqueratosis folicular, atriquia y puede producirse fotofobia grave con queratitis vascularizantes.
Síndrome de Sjӧgren Larsson: con herencia AR, se caracteriza por hiperqueratosis laminar, paraplejía espástica, retraso mental y puntos blancos brillantes en la retina.
Enfermedad de Refsum: con herencia AR, presenta la tétrada de retinitis pigmentaria, neuropatía periférica, ataxia cerebelosa y sordera progresiva.
Síndrome de Conradi-Hϋnermann-Happle (CDPX2): con herencia dominante ligada al cromosoma X, presenta eritema llamativo, condrodisplasia puntiforme con hipoplasia esquelética asimétrica, cataratas y baja estatura.
Síndrome de Gaucher tipo 2: herencia AR. Nace con membrana colodión leve, luego queda descamación generalizada o la piel es normal, hidropesía fetal, deterioro neurológico progresivo y hepatoesplenomegalia.14
En la ictiosis vulgar las características histopatológicas suelen ser la hiperqueratosis ortoqueratósica moderada y compacta, tapones córneos, granulosa normal o aumentada y adelgazamiento de la capa de Malpigui, presencia de pequeños infiltrados linfocíticos perivasculares; puede haber hipotrofia de glándulas sebáceas y sudoríparas.1
Con todos estos elementos se determinó que el cuadro ictiosiforme de la paciente se correspondía más con el de la ictiosis vulgar, corroborado por el estudio histopatológico; pero las manifestaciones de hipermovilidad articular e hiperextensibilidad de la piel no se correspondían con ninguna de las manifestaciones en la clasificación de las ictiosis. Se pensó entonces en la asociación con el síndrome de Ehlers Danlos.
El síndrome de Ehlers Danlos resulta una genodermatosis que afecta el tejido conectivo, fundamentalmente el de la piel. Presenta hiperextensibilidad cutánea, y de las articulaciones dadas por hipermovilidad articular, puede afectar otras estructuras del sistema osteomioarticular y otros órganos como los vasos sanguíneos y el ojo.
Desde 2017 se reconocieron 13 subtipos en la clasificación internacional del síndrome de Ehlers Danlos.15
Forma clásica: con herencia AD, presenta hiperextensibilidad de la piel, estrías, cicatrices atróficas e hipermovibidad articular.
Síndrome de Ehlers Danlos similar al clásico: con herencia AR, se caracteriza por hipermovilidad articular, luxaciones frecuentes, dolor crónico articular y piel normal.
Forma vascular: con herencia AD, presentan defectos en la agregación plaquetaria con tendencia a sangramientos, piel translúcida, acrogeria y aneurisma. No se asocia con hiperextensibilidad de las grandes articulaciones; la hiperlaxitud se limita a las pequeñas articulaciones de las manos.
Forma valvular cardíaca: con herencia AR, presentan problemas cardiovasculares progresivos, hiperextensibillidad de la piel, cicatrices atróficas e hipermovilidad generalizada de articulaciones.
Forma sifoescoliótica: con herencia AR, se caracteriza por hipotonía muscular desde el nacimiento, hiperextensibilidad articular, sifoescoliosis y lesiones oculares.
Síndrome de córnea frágil: con herencia AR. Suelen presentar fractura de la córnea, queratoconos y escleras azules.
Forma tipo dermatosparaxis: con herencia AR, se caracteriza por presentar fragilidad de la piel y cutis laxo.
Forma tipo artrocalasia: con herencia AD, presentan hiperextensibilidad de la piel, corta estatura con dislocación congénita de algunas articulaciones.
Forma espondilodisplásica: con herencia AR, se caracteriza por corta estatura con hipotonía muscular desde la infancia y extremidades arqueadas.
Forma musculocontractural: con herencia AR, se presentan contracturas congénitas múltiples, facie característica, hiperextensibilidad de la piel y cicatrices atróficas.
Forma miopática: con herencia AD o puede ser AR, presentan hipotonía muscular desde edades tempranas con atrofia, hipermovilidad de articulaciones distales y contractura de articulaciones proximales.
Forma hipermóvil: con herencia AD, se caracteriza por hiperlaxitud articular generalizada, piel hiperextensible, suave, aterciopelada; hernias y prolapsos, luxación o inestabilidad articular recurrente, aracnodactilia y dolor crónico articular o generalizado.
Forma periodontal: con herencia AD, caracterizada por hiperextensibilidad de la piel, fragilidad de la mucosa y periodontitis.15,16
Existen criterios diagnósticos, según las diferentes formas clínicas. En la forma clásica se debe tener en cuenta la presencia obligatoria del criterio mayor: piel hiperextensible con formación de escaras atróficas o la presencia de hiperlaxitud articular; acompañado de dos o más criterios menores como: piel tersa, aterciopelada, magulladuras, pseudotumores moluscoides, esferoides subcutáneos, hernias, complicaciones de la hiperlaxitud (luxaciones, pie plano flexible), hipotonía muscular, historia familiar positiva.15
El primer paso para la identificación del riesgo genético es conocer la historia genética familiar a través del árbol genealógico como principal herramienta profesional en el campo de la genética médica.17) El análisis del árbol genealógico permitió determinar los patrones de herencia y guiar el diagnóstico que planteó la concomitancia en una misma paciente de una ictiosis vulgar (confirmado con los elementos histopatológicos de la biopsia de piel, y se descartó la posibilidad de síndromes genéticos complejos como los síndromes ictiosiformes) asociada al síndrome de Ehlers Danlos de tipo clásico (cumplía con los criterios diagnósticos para esta forma clínica).
En la literatura revisada, solo aparecieron publicaciones de casos con algunas formas de ictiosis, fundamentalmente de ictiosis laminares,4,12 y casos con formas clínicas poco frecuentes del síndrome de Ehlers Danlos,10,18 no se localizaron publicaciones de casos en los que coexistieran ambas genodermatosis en el mismo paciente.
Aunque no es frecuente, se documentan en la literatura algunos casos en el que coincidieron más de una enfermedad genética, por ejemplo, el caso de un paciente que presentaba neurofibromatosis tipo I asociada con el síndrome de Klippel-Trenaunay.19
Se concluye que es infrecuente encontrar en un mismo paciente dos enfermedades genéticas, lo cual hace muy difícil el diagnóstico, así como su atención. Se determinó el diagnóstico de ambas genodermatosis con el análisis del árbol genealógico familiar como herramienta fundamental en el diagnóstico de enfermedades genéticas.

