










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Obstetricia y Ginecología
On-line version ISSN 1561-3062
Rev Cubana Obstet Ginecol vol.43 no.1 Ciudad de la Habana Jan.-Mar. 2017
PRESENTACIÓN DE CASO
Éxitus materno tras cesárea en gestante con anomalía de Taussig-Bing y síndrome de Eisenmenger
Maternal Exitus after Cesarean Section in Pregnant Women with Taussig-Bing Anomaly and Eisenmenger Syndrome
Manuel Pantoja Garrido,I Zoraida Frías Sánchez, II Azahara Romero Lozano,I Pilar Guadix Martínez,II María Jesús Parra Fernández II, Isabel Corrales Gutiérrez II
I Unidad de Gestión Clínica de Obstetricia y Ginecología del Hospital Universitario de Jerez de la Frontera. Cádiz, España.
II Unidad de Gestión Clínica de Obstetricia y Ginecología del Hospital Universitario Virgen Macarena de Sevilla. España.
RESUMEN
La anomalía de Taussig-Bing es una cardiopatía congénita cianosante caracterizada por la dextrotransposición de grandes vasos. Esta produce una doble salida arterial desde el ventrículo derecho, asociado a una comunicación interventricular. Este cuadro puede generar una hipertensión pulmonar secundaria al aumento de las resistencias vasculares y un flujo reverso cardiaco, conocido como síndrome de Eisenmenger. Normalmente, se presenta antes de la pubertad, aunque en ocasiones, puede debutar en la vida adulta, progresando durante dicha etapa. Clínicamente, se caracteriza por rasgos crónicos, como las acropaquias, la disnea, la sensación de cansancio o la cianosis. El diagnóstico de este tipo de cardiopatías se basa en la clínica y en las pruebas de imagen, preferentemente en el estudio ecocardiográfico fetal o durante la edad pediátrica. El tratamiento de elección es la corrección quirúrgica de las malformaciones cardiacas, siendo preferente la rectificación de la salida de la aorta y el cierre de la comunicación interventricular. El pronóstico depende del grado de hipertensión pulmonar, del momento del diagnóstico y de la corrección quirúrgica precoz. A edades tempranas se obtiene un mejor resultado, aunque las tasas de mortalidad alcanzan 50 % en algunos casos, incluso tras una corrección quirúrgica óptima. La gestación no está recomendada en pacientes que padecen dicha patología, la cual se ha contraindicado, según algunos estudios, en ausencia de tratamiento adecuado. Así pues, describimos un caso en el que una gestante con una anomalía Taussig-Bing sufre una atonía uterina y un posterior paro cardiorrespiratoria tras el parto, realizado mediante cesárea electiva, tras la que la paciente falleció.
Palabras clave: anomalía Taussig-Bing; doble salida ventrículo derecho; transposición grandes vasos; enfermedades cardiovasculares; síndrome Eisenmenger; embarazo.
ABSTRACT
The Taussig-Bing anomaly is a congenital cyanosis characterized by the dextrotransposition of large vessels. It produces a double arterial exit from the right ventricle, associated with an interventricular communication. This may lead to pulmonary hypertension secondary to increased vascular resistance and a cardiac reverse flow, known as Eisenmenger syndrome. Generally, it occurs before puberty, although occasionally, it can debut in adults, progressing during that stage. It is clinically characterized by chronic features, such as acropachies, dyspnea, tiredness or cyanosis. The diagnosis of this type of heart disease is based on clinical exam and imaging tests, if at all possible in the fetal echocardiographic study or during the pediatric age. The surgical correction of cardiac malformations is the treatment of choice, modifying the aortic exit and closing of ventricular septal defect. The prognosis depends on the degree of pulmonary hypertension, the time of diagnosis, and the early surgical correction. Better outcome is obtained at early ages, although mortality rates reach 50% in some cases, even after optimal surgical correction. Pregnancy is not recommended in patients suffering from this disease. Some studies contraindicate pregnancy in absence of proper treatment. Thus, we describe a case of a pregnant woman with a Taussig-Bing anomaly, who suffered uterine atony and a subsequent cardiorespiratory arrest after delivery. Elective cesarean section was performed. This patient died.
Keywords: Taussig-Bing anomaly; double exit right ventricle; transposition of great vessels; cardiovascular diseases; Eisenmenger syndrome; pregnancy.
INTRODUCCIÓN
La anomalía Taussig-Bing es una rara malformación cardiaca, descrita por primera vez en 1949 por Helen B. Taussig y Richard J. Bing, que representa entre 1-5 % del total de cardiopatías congénitas. 1 Consiste en la transposición de la salida de la aorta desde el ventrículo derecho y la malposición de la arteria pulmonar, asociada a una comunicación interventricular subpulmonar. Puede asociarse a una coartación aórtica, lo que empeora significativamente el pronóstico clínico.2,3 Es una patología muy compleja que supone un desafío quirúrgico importante, en gran medida, debido a las anomalías asociadas a este síndrome, entre las que destacan significativamente los diferentes grados de salida de la aorta del ventrículo derecho y la obstrucción del arco aórtico causado por el desarrollo del tabique ventricular.1,2,3 Es una cardiopatía cianosante asociada, en un alto porcentaje de casos, a hipertensión pulmonar grave con flujo bidireccional (conocido como Síndrome de Eisenmenger) que, clínicamente, se evidencia como disnea a mínimos esfuerzos y cianosis.4,5 En estos casos, la extrema gravedad del cuadro puede llevar a una hipertrofia ventricular derecha con un deterioro de la propia reserva ventricular.6 Fue descrito por primera vez en el año 1897 por el doctor Víctor Eisenmenger. Actualmente, esta patología alcanza una incidencia aproximada, en pacientes con cardiopatías congénitas del 3 %. Se trata de un síndrome con una alta mortalidad en gestantes. Se objetiva -en algunas series- cifras de hasta el 30 %, relacionado con fenómenos tromboembólicos, hipovolemia y estados hipertensivos como la preeclampsia.4,5 Todo esto hace que sea considerada una patología de alto riego obstétrico para la gestante, con un elevado riesgo de muerte materna, contraindicando el embarazo en pacientes con esta anomalía.4
Actualmente, dentro de los procedimientos terapéuticos para el tratamiento de estas infrecuentes cardiopatías, se han conseguido tasas de éxito significativas en la reducción del porcentaje de reintervenciones, mortalidad y complicaciones neo-aórticas, particularmente en pacientes que habían sido sometidos a procedimientos paliativos, como la colocación de bandas en la arteria pulmonar antes de la reparación definitiva.1,6,7 Así mismo, es importante reportar los casos relacionados con este tipo de patología, ya que es una enfermedad muy poco frecuente. En la literatura se encuentran menos de 200 publicaciones relacionadas (que alcanza cifras casi inexistentes si se trata de gestantes), y con una alta tasa de mortalidad sin el diagnóstico ni el tratamiento correctivo adecuado.
A continuación, describimos el caso de una gestante de 23 años diagnosticada de una malformación de Taussig-Bing, asociada a síndrome de Eisenmenger durante su segundo embarazo. Tras un estricto seguimiento de la gestación, el servicio de Cardiología indicó la finalización mediante cesárea electiva, posteriormente se produjo un cuadro de inestabilidad hemodinámica con parada cardiorrespiratoria y el éxitus de la paciente a las 5 horas de la intervención.
PRESENTACIÓN DE CASO
Exponemos el caso de una paciente de 24 años de edad, originaria de Rumania, que acude al programa de seguimiento de embarazo. Refiere como antecedentes personales, un parto eutócico a las 38 semanas de gestación tras embarazo no controlado, con buenos resultados perinatales, aunque el feto presentó un percentil 1 de peso al nacer (2230 gr).
Fue derivada a la Unidad de Alto Riesgo obstétrico por presentar una cardiopatía congénita no documentada correctamente; la cual, refiere que fue diagnosticada en su país de origen, pero de la que no aporta ningún informe. Refiere antecedentes de varios episodios de crisis hipertensivas y de taquicardia espontáneas; las cuales no están asociadas a esfuerzo, acompañada de sensación de fatiga y disnea.
Los controles ecográficos que se realizaron a las 12 y 20 semanas de gestación se encuentran dentro de la normalidad, sin presencia de alteraciones morfológicas y evolucionando de forma normal. El seguimiento del embarazo se desarrolló adecuadamente, pero en la ecografía morfológica de las 32 semanas, se objetivó que el feto presentó un bajo peso para su edad gestacional (1581 gr, (percentil 9), sin alteraciones de la placenta o del líquido amniótico.
Se volvió a valorar pasados 7 días, incluyendo un estudio ecográfico avanzado, en el que destacó la presencia de un crecimiento intrauterino restringido (CIR). Mantuvo un percentil 3 de peso para su edad gestacional, sin alteraciones en el flujo Doppler. Dados los hallazgos, la edad gestacional y la comorbilidad de la paciente, se continúa con un estricto control tanto obstétrico como cardiológico. Una vez valorada por el servicio de Cardiología, se exploró detenidamente a la gestante y se le solicitaron diversas pruebas complementarias.
La paciente presentó una saturación de oxigeno del 70 % y un soplo pansistólico en borde esternal, irradiado a foco tricuspídeo, con una frecuencia cardiaca de 95 latidos por minuto (lpm). Las cifras tensionales se encuentran dentro de la normalidad.
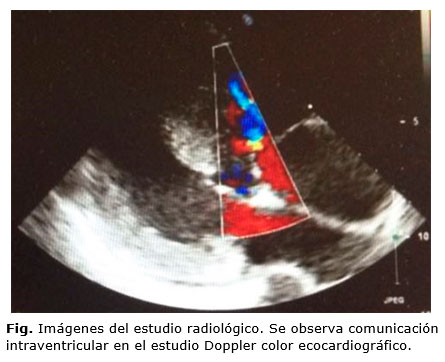
Se realizó un electrocardiograma (ECG) que presentó ritmo sinusal, crecimiento de la onda P en aurícula derecha, eje desviado a la izquierda y crecimiento biventricular. En la ecocardiografía, además, se objetiva una cardiopatía congénita compleja cianótica con doble salida de ventrículo derecho y dextrotransposición de grandes arterias, insuficiencia grado II de la válvula pulmonar, hipertrofia biventricular, comunicación interventricular subpulmonar e hipertensión pulmonar (Síndrome de Eisenmenger) (Fig.). Todo ello se acompaña de desaturación arterial severa, con clase funcional grado II y riesgo WHO (evaluación riesgo cardiorrespiratorio preoperatorio) grado IV. Finalmente, y ante los hallazgos descritos, se diagnostica de anomalía de Taussig-Bing asociado a Síndrome de Eisenmenger. Como tratamiento, se estableció oxigenoterapia, reposo relativo durante el resto de la gestación, heparina de bajo peso molecular, profilaxis de endocarditis a las 38 semanas y finalización mediante cesárea electiva.
En la última visita obstétrica, se volvió a valorar a la gestante y se comprobó que el feto continuaba con un peso estimado en un percentil 4,2308 gr, con estudio de flujometría normal y registro cardiotocográfico sin alteraciones. En esa misma consulta, se informó detalladamente a la paciente de la intervención que se le va a realizar. Se le ofertó bloqueo tubárico bilateral como método anticonceptivo definitivo, el cual aceptó a pesar de la gravedad de un embarazo con la patología de base que presenta. Durante la intervención, nace un feto con Apgar 10/10/10 y pH de arteria umbilical normal, continuando con la cesárea sin incidencias. Sin embargo, tras el alumbramiento dirigido, la paciente sufre una descompensación hemodinámica y un paro cardiorrespiratorio que requiere medidas de reanimación cardiopulmonar avanzada. Se realizó masaje cardiaco y administración intravenosa de sueroterapia, dobutamina, noradrenalina e infusión de bolos de fenilefrina, tras lo cual, se consiguió una recuperación hemodinámica y de las cifras de saturación arterial de oxígeno (SO2) adecuadas.
En ese momento, el hemograma y la gasometría arterial presenta un ph de 7,20, unos niveles de bicarbonato de 21,7 (HCO3), una presión parcial de dióxido de carbono (pCO2) de 50,6 mmHg, una presión parcial de oxígeno (pO2) de 31,4 mmHg y una concentración de hemoglobina de 13 gr/dl.
La gravedad de la situación obligó a finalizar la cesárea en el menor tiempo posible, por lo que se terminó la intervención sin realizar el bloqueo tubárico. Al finalizar la intervención, la paciente presentó un nivel de conciencia y un estado neurológico conservado. No obstante, mantuvo cifras tensionales de 200/100 mmHg y una SO2 del 75 %. Se realizó profilaxis de hemorragia postparto, con prostaglandinas intrarrectales y se consensúa con el servicio de Anestesia y de Intensivistas, el traslado de la paciente a la Unidad de Cuidados Intensivos. Una vez en la UCI, la paciente comienzó a mostrar signos de inestabilidad hemodinámica, con taquicardia, taquipnea, descenso de la SO2 al 45 % y sangrado vaginal en cuantía abundante que hace descender la hemoglobina de 13 gr/dl a 11 gr/dl, por lo que se decide avisar al equipo de ginecología de guardia.
Durante la exploración, se confirmó una metrorragia llamativa, con atonía uterina marcada que requiere administración de fármacos uterotónicos con oxitocina intravenosa, metilergometrina intramuscular, carbetocina y masaje uterino continuo. El episodio se estabilizó a las dos horas. Poco después, volvió a presentar un cuadro semejante, transfundiendo varios concentrados de hematíes, plasma, plaquetas, complejo protrombínico, Factor VIIa y fibrinógeno. Ante la progresión del episodio, se colocó un balón de taponamiento intrauterino, que no logró el cese del sangrado; así que se decidió realizar una laparotomía exploradora.
En la intervención, destacó un hemoperitoneo masivo con una atonía uterina marcada; por lo cual se le realizó una histerectomía subtotal de forma urgente. No obstante, y pese a todas las medidas terapéuticas realizadas, se produjo un importante choque hipovolémico de origen hemorrágico, que finalizó en un paro cardiorrespiratorio y el fallecimiento de la paciente a las 5 horas de la cesárea.
DISCUSIÓN
Las malformaciones cardiacas constituyen un grupo de patologías importante dentro de las anomalías congénitas, debido a sus múltiples variantes y combinaciones posibles. En la actualidad, continúan siendo un reto diagnóstico y terapéutico -en especial- aquellas cardiopatías complejas en las que, por su baja incidencia, existen pocos grupos experimentados en el manejo de ellas.3 El síndrome de Taussig-Bing es una cardiopatía congénita extremadamente infrecuente, en la que tanto la arteria pulmonar como la aorta se originan predominantemente en el ventrículo derecho (DORV). Está asociada a un defecto del septo interventricular a nivel subpulmonar, que enfatiza el acabalgamiento de la misma sobre defecto septal, como parte integral de la malformación.3,8 La incidencia total de cardiopatías durante el embarazo oscila entre 0,4 % y 4,1 %, mientras que la probabilidad de presentar una anomalía de Taussig-Bing es extremadamente baja; se desarrolla aproximadamente en 0,5-0,8 casos por cada 10,000 nacimientos.8 Los factores pronósticos más importantes en este grupo de trastornos están relacionados con la clase funcional de estos (NYHA) y las manifestaciones clínicas acompañantes, como el grado de disnea, fatigabilidad o cianosis.8
Las gestantes que padecen estas anomalías cardiacas complejas, sufren importantes trastornos cardiovasculares durante el embarazo y el parto, por lo que se considera una patología de alto riesgo obstétrico. Fisiológicamente, la gestación provoca un aumento del gasto cardíaco de entre 30-50 %, un aumento en el volumen sanguíneo del 40-50 % y del consumo de oxígeno de casi 20 %, factores que pueden producir grandes descompensaciones en pacientes con patología cardiaca de base asociada.6 Durante el trabajo de parto, se produce un aumento del gasto cardiaco secundario a un incremento de la volemia intravascular, cercana a los 500 ml de sangre con cada contracción uterina. Esto puede derivar en un cuadro de insuficiencia cardiaca en pacientes con este síndrome, ya que la malformación cardiaca impide distribuir correctamente ese aumento de volumen.6
Nuestra paciente no tuvo contracciones, pero sufrió una desestabilización hemodinámica importante como consecuencia de la cirugía y el alumbramiento placentario, que desencadenó un cuadro de insuficiencia cardiaca. La comunicación interventricular asociada a este síndrome produce un alto flujo pulmonar que puede generar un flujo reverso cardiaco, disminuyendo así la correcta oxigenación sanguínea y derivando en un aumento de las resistencias vasculares e hipertensión pulmonar (Síndrome de Eisenmenger), como en nuestro caso. Los casos más severos pueden desembocar en fallo cardiaco por aumento de la poscarga en el ventrículo derecho. Cuando se presenta otras alteraciones asociadas a estos síndromes congénitos (como coartación de aorta), se agrava, lo cual produce una hipertrofia ventricular derecha con deterioro de la reserva ventricular.4,5,6,7
La caída de la resistencia vascular sistémica aumenta la derivación derecha-izquierda e incrementa la cianosis, y las alteraciones hemodinámicas durante el trabajo de parto y el periodo de expulsivo (incluyendo la pérdida hemática); esto puede precipitar un colapso cardiovascular importante. La alta mortalidad asociada a estos trastornos, suele desarrollarse con frecuencia durante el trabajo de parto, expulsivo y puerperio inmediato. Aunque la adición de terapias vasodilatadoras pulmonares ha mejorado las tasas de supervivencia en las gestantes con síndrome de Eisenmenger, no debe obviarse que el resultado para muchas de estas pacientes sigue siendo -a menudo- fatal.
Actualmente, un alto porcentaje de las cardiopatías congénitas mayores se diagnostican durante el estudio morfológico ecográfico prenatal. Estos diagnósticos permiten iniciar protocolos terapéuticos muy precoces, y consensuar la elección sobre la evolución de la gestación, en fases muy tempranas. La imagen ecográfica convencional en 2 dimensiones (2D) aún es limitada para algunas aplicaciones, a pesar de las mejoras en la resolución y en el equipamiento de los dispositivos. Debido a la falta de información sobre la profundidad de campo, la definición precisa de las relaciones intracardiacas, es un reto.
La introducción de técnicas de ecocardiografía fetal en 3D, tales como la tecnología STIC (Imagen de correlación espacio-temporal), permite que el volumen ecográfico sea visto en proyecciones ortogonales y así, lograr una evaluación detallada, que mejora la visualización de la anatomía, llevando a una predicción precisa del tipo de reparación quirúrgica que se puede llevar a cabo en el futuro.9 La ecocardiografía fetal permite el diagnóstico prenatal de muchas anomalías cardiacas estructurales y funcionales. Es importante relacionar la presencia de bradicardia fetal durante el examen ecográfico, ya que puede ser un indicador adicional de una anomalía cardiaca subyacente grave, con el aumento de la translucencia nucal en el estudio morfológico fetal (indicador de cromosomopatías relacionadas con anomalías cardiacas congénitas).10 Por otro lado, en pacientes pediátricos, la angiografía por sustracción digital (DSA) se consideraba la prueba de imagen inicial de elección para el diagnóstico, seguimiento y planificación terapéutica de las DORV.
Sin embargo, este es un procedimiento invasivo que conlleva riesgos asociados importantes, con complicaciones relacionadas con el cateterismo y con la alta dosis de radiación a la que son sometidos este tipo de pacientes. Por ello, la asociación de esta técnica durante la última década, a la ecocardiografía tridimensional ha sido considerada como un procedimiento de diagnóstico por imagen de primera línea, para pacientes pediátricos con DORV.8,11,12
Actualmente, se está dando importancia a la resonancia magnética nuclear como una técnica de imagen prometedora en el diagnóstico tridimensional de malformaciones cardiacas, debido sobre todo a su exactitud y naturaleza libre de radiación.9 Nuestra paciente no fue sometida a ninguna de estas pruebas durante la infancia, por lo que su diagnóstico se basó en el estudio ecocardiográfico realizado por el servicio de Cardiología durante la gestación.
El tratamiento definitivo de este tipo de cardiopatía congénita es principalmente quirúrgico, e implica la reparación de la comunicación interventricular y la corrección de la trasposición de grandes arterias asociada en la primera infancia. Cuando los pacientes sobreviven y alcanzan la edad adulta sin haber sido sometidos a cirugía, acaban desarrollando hipertensión pulmonar grave con flujo bidireccional, que se manifiesta clínicamente como disnea y cianosis. Dichos síntomas no asociados a esfuerzo, fueron referidos por nuestra paciente en la primera visita gestacional.
Intraoperatoriamente, los objetivos durante el procedimiento son la optimización de la precarga, estabilización de la frecuencia cardiaca y de la resistencia vascular pulmonar (PVR), manteniendo la resistencia vascular sistémica en la normalidad o ligeramente elevada.
El abordaje quirúrgico consiste en la conexión del ventrículo izquierdo a la circulación sistémica, manteniendo la circulación pulmonar asociada a las cavidades cardiacas derechas. Para ello, existen diferentes técnicas quirúrgicas que permiten obtener buenos resultados terapéuticos y pronósticos, como el Switch arterial, la operación de Damus-Rastelli o el túnel interventricular de Kawashima.3,12,13,14 El Switch arterial o corrección anatómica de la transposición de las grandes arterias, es el tratamiento de elección en los casos de doble salida ventricular, y consiste en la realización de un bypass coronario con interposición de injertos vasculares de politetrafluoroetileno. De los pacientes con anomalía de Taussig- Bing 35 % - 50 % son candidatos a este tipo de cirugía.
La continuidad entre el ventrículo derecho y el tronco pulmonar distal se restablece mediante la interposición de un conducto extracardiaco (intervención de Yasui (Damus-Rastelli)).3 Está indicada en aquellos pacientes en los que el Switch arterial no es una opción recomendada. Por último, la técnica del túnel interventricular de Kawashima, tiene la ventaja de preservar la válvula aórtica nativa y evitar la disección coronaria. Sin embargo, en la mayoría de los casos, es necesaria la realización de una ventriculotomía derecha para realizar una resección del cono subaórtico y dejar sin obstrucción de salida el túnel intraventricular. La ventriculotomía derecha debe cerrarse con un parche adicional para evitar la obstrucción secundaria del flujo de salida subpulmonar.2,7,13,14
Las principales complicaciones derivadas de este tipo de intervenciones quirúrgicas son -por su complejidad- las hemorragias posoperatorias, el síndrome de bajo gasto, el fallo biventricular, las arritmias o el fallo renal agudo. Por ello, hay que realizar una vigilancia estrecha del paciente durante el posoperatorio inmediato, para hacer un diagnóstico temprano de estas complicaciones.
En la literatura existen pocos casos descritos de corrección total de la malformación de Taussig-Bing, por ser una cardiopatía poco común, cuyo tratamiento óptimo tiene una alta tasa de mortalidad, con un alto índice de hemorragias, insuficiencia cardiaca y alteraciones de la coagulación.3
Nuestra paciente no fue intervenida quirúrgicamente de la cardiopatía congénita (de hecho, no era conocedora realmente de la gravedad de su enfermedad), lo que incrementa exponencialmente la morbimortalidad del cuadro. Aunque su sintomatología clínica era leve y presentó disnea de pequeños y/o moderados esfuerzos y fatiga, los datos ecocardiográficos revelaron una importante afectación de la morfología y función cardiaca, bastante evolucionada y que comportaba un compromiso vital muy grave.
Existen mínimas referencias sobre el manejo de la anomalía de Taussig-Bing en gestantes, ya que como indicamos anteriormente, son extremadamente infrecuentes los casos de pacientes con estas alteraciones que lleguen a la edad reproductiva sin recibir tratamiento. La prostaciclina intravenosa (epoprostenol), el sildenafilo, el tadalafilo, así como iloprost nebulizado e intravenoso, o el óxido nítrico inhalado, pueden considerarse terapias periparto útiles. Todos estos tratamientos están dirigidos a conseguir una reducción de las resistencias pulmonares y una estabilización de la función ventricular derecha; aun así, el porcentaje de pérdidas fetales es elevado, con altas tasas de abortos espontáneos y de retrasos del crecimiento intrauterino4,5 (como el que desarrollo durante la primera gestación).
La gestación en estas pacientes es un tema muy controvertido, sin evidencia suficiente para establecer un protocolo único. Así pues, en los casos de mayor complejidad, es fundamental un asesoramiento prenatal, ya que parece existir un consenso al contraindicar la gestación.
No existen protocolos establecidos sobre la vía más adecuada para el parto. Sin embargo, en algunos artículos que hacen referencia a esta disyuntiva, la elección suele ser la cesárea electiva programada asociada a técnicas analgésicas epidurales, ya que parece reducir el retorno venoso y aliviar la carga cardiaca.6,8 No obstante, hay series que describen la tasa de mortalidad tras el parto vaginal cercana al 35 %, la cual aumenta hasta 75 % si la finalización es mediante cesárea.5 Es imprescindible considerar este tipo de gestaciones de alto riesgo obstétrico, se requiere un seguimiento exhaustivo y multidisciplinar. Nuestra paciente no conocía la gravedad de su enfermedad, por lo que al obtener un diagnóstico definitivo de su cardiopatía, ya se encontraba embarazada por segunda vez, así que en este caso el asesoramiento previo no fue el adecuado. La elección de la vía del parto se considera un tema controvertido, puesto que la paciente había tenido un parto eutócico anterior con buenos resultados tanto maternos como fetales.
El pronóstico a largo plazo en este tipo de patologías depende de múltiples factores, como el momento del diagnóstico, el tipo de tratamiento recibido o la patología de base que tenga la paciente. Aun así, el pronóstico no es muy esperanzador, ya que clasificaciones especializadas como la escala RASCH-1, nos indican que la tasa de mortalidad alcanza 50 % en pacientes con una corrección quirúrgica total de la cardiopatía.3 Por ello, debemos considerar que nuestra paciente tenía un pronóstico funesto, independientemente de las medidas terapéuticas que se llevaran a cabo, debido al diagnóstico tardío, a la ausencia de tratamiento de su cardiopatía de base, y a la gestación como responsable de la desestabilización de su estado hemodinámico.
CONCLUSIONES
La anomalía de Taussig-Bing es una cardiopatía congénita muy infrecuente, que presenta una gran repercusión hemodinámica, en ocasiones, asociada a síndrome de Eisenmenger. El número de casos publicados es escaso y, si se trata de gestantes con dicha patología, la cifra es casi inexistente dada la alta mortalidad que conlleva. La clínica se deriva de la hipertensión pulmonar crónica que provoca disnea, cianosis, acropaquias o sensación de cansancio. El diagnóstico suele ser prenatal, aunque existen casos que pueden ser estudiados en la vida adulta. El tratamiento de esta patología es principalmente quirúrgico e intenta lograr una corrección de las malformaciones asociadas. El pronóstico es variable, en función del diagnóstico precoz, del tratamiento adecuado de las malformaciones asociadas y de la presencia de hipertensión pulmonar severa. La gestación está contraindicada en el caso de síndrome de Eisenmenger, dada la alta mortalidad de las gestantes con esta patología de base. Es fundamental realizar un diagnóstico preciso y precoz, si es que no se ha detectado de forma prenatal, para poder ajustar el tratamiento lo antes posible y permitir un pronóstico más favorable. En caso de gestación, estas pacientes deben tener un seguimiento exhaustivo y multidisciplinar en unidades de alto riesgo obstétrico, junto a equipos de cardiología experimentados en este tipo de síndromes. Se pone especial énfasis en la elección de la vía del parto, ya que no existe un consenso claro sobre que vía puede ser la recomendada, debido a la alta mortalidad asociada a ambas.
Conflicto de intereses
Los autores no declaran tener conflictos de intereses.
REFERENCIAS BIBLIOGRÁFICAS
1. Konstantinov IE. Taussig-Bing Anomaly. Tex heart Inst J. 2009;36:380-585.
2. Caffarena Calvar JM, Bautista V and Serrano F.R. Transposiciones complejas y corazón de Taussing- Bing. Lecciones aprendidas. Cir Cardiov. 2007;14(2):111-8.
3. Garza Alatorre AG. Corrección completa de defecto Taussing Bing técnica Damus Rastelli: Reporte de Caso. Rev Mex Cardiol. 2015;26(3):122-5.
4. Warnes CA. Pregnancy and Delivery in Women with congenital heart disease. Circulation Journal. 2015;7:1416-21.
5. Rathod S, Kumar Samal S. Successful Pregnancy Outcome in a case of Eisenmenger Syndrome: A rare case report. J Clin Diagn Res. 2014;8(10): OD 08-OD09.
6. Mishra G, Bindu Nagella A, Parthasarathy S and Vivek B. Segmental epidural anesthesia for cesarean section in a parturient with uncorrected Taussig-Bing anomaly with transposition of the great arteries physiology. Anesth Essavs Res. 2015;9(3):408-10.
7. Hayes DA Primary Arterial Swith Operacion as a Strategy for total Correction of Taussig-Bing Anomaly. Circulation. 2013;10:194-8.
8. Gu J, Cai Y, Lui B and Lv S. Anesthetic management for cesarean section in a patien with uncorrected doublé outlet right ventricle. Poslepy Kardiol Interwecyjnej. 2016;12(3):267-270.
9. Zidere V, Pushparajah K, Allan LD, Simpson JM. Three-dimensional fetal echocardiography for prediction of postnatal surgical approach in double outlet right ventricle: a pilot study. Ultrasound Obstet Gynecol. 2013;42(4):421-5.
10. Baschat A, Gembruch U, Knöpfle G and Hansmann M. First-trimester fetal heart block: a marker for cardiac anomaly. Ultrasound Obstet Gynecol. 1999;14:311-4.
11. Shi K. Assessment of Double Outlet Right Ventricle Associated with Multiple Malformations in Pediatric Patients Using Retrospective ECG-Gated Dual-Source Computed Tomography. PLoS One. 2015;10(6):e013-0987.
12. Smith S. Double-outlet right ventricle: an antenatal diagnostic dilema. Ultrasound Obstet Gynecol. 1999;14:315-9.
13. Khairi P. Cardiovascular outcomes after the arterial switch operation for D-transposition of the great arteries. Circulation. 2013;127(3):331-9.
14. Blume ED. Evolution of risk factors influencing early mortality of the arterial switch operation. J Am Coll Cardiol. 1999;33(6):1702-9.
Recibido: 22 de agosto de 2016.
Aprobado: 24 de septiembre de 2016.
Manuel Pantoja Garrido. Unidad de Gestión Clínica de Obstetricia y Ginecología del Hospital Universitario de Jerez de la Frontera. Cádiz, España.

