










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Salud Animal
Print version ISSN 0253-570XOn-line version ISSN 2224-4700
Rev Salud Anim. vol.38 no.1 La Habana Jan.-Apr. 2016
ORIGINAL ARTICLE
Standarization of a SYBR Green-I based real-time RT-PCR assay for detection of bluetongue virus in centinel animals
Estandarización de un ensayo de RT-PCR en tiempo real basado en SYBR Green I para la detección del virus de la lengua azul en animales centinelas
Ana María Acevedo, Armando Vega, Yoandry Hinojosa, Ana M. Lazo, Damarys Relova, Liani Coronado, María T. Frías, Carmen L. Perera
Animal Virology Group, National Center for Animal and Plant Health (CENSA), Apartado 10, San José de las Lajas, CP 32 700, Mayabeque, Cuba. E-mail: acevedo@censa.edu.cu.
ABSTRACT
Bluetongue (BT) is an insect-transmitted viral disease of ruminant species that primarily affects sheep. This disease is caused by a virus (BTV) of the genus Orbivirus within the family Reoviridae. Twenty-six distinct serotypes of the virus have been identified to date. The rapid dissemination of the virus and the variability of the clinical signs merit the development of swift and accurate BTV detection methods, which can assist in disease control. SYBR Green-based real-time reverse transcription-PCR (rRT-PCR) assay may be an ideal method to detect rapidly the BTV genome in the blood of animals. In this study, a rRT-PCR coupled to melting curve analysis was described and compared with the nested reverse transcription-polymerase chain reaction (nRT-PCR) reported in the OIE Manual. The specificity and sensitivity of both methods were evaluated using BTV-4 RNA extracted from tenfold serially diluted tissue culture medium virus and blood (starting from 104.5 TCID50/ml). The detection limit of each test in tissue culture medium was 100.5 TCID50/ml and in blood was 101.5 TCID50/ml (rRT-PCR) and 100.5 TCID50/ml (nRT-PCR). Melting curve analysis showed that the rRT-PCR yielded curves of amplification with specific melting curves (Tm = 84°C ± 0.5°C) and absence of non-specific amplifications. The diagnostic sensibility of clinical samples from a calf under controlled conditions was evaluated. These results showed that the SYBR Green I-based real-time PCR assay is rapid, sensitive, and equally specific in the diagnosis of BT in BTV-affected animals.
Key words: bluetongue virus, nested RT-PCR, SYBR Green I-based real-time RT-PCR assay.
RESUMEN
La lengua azul es una enfermedad viral de especies de rumiantes que se transmite por insectos, y que primariamente afecta a los carneros. La enfermedad es causada por un virus (BTV) que pertenece al género Orbivirus de la familia Reoviridae. Se han identificado 26 serotipos diferentes de este virus hasta la fecha. Debido a la rápida diseminación del virus y a la variabilidad de los signos clínicos, se necesita el desarrollo de métodos de detección de BTV rápidos y precisos, los cuales pueden ayudar en el control de la enfermedad. Un ensayo de RT-PCR en tiempo real (rRT-PCR), basado en SYBR Green-I, puede ser un método ideal para detectar rápidamente el genoma de BTV en la sangre de los animales. En este estudio se describe un análisis de curvas de disociación acoplado al rRT-PCR y se compara con el ensayo de RT-PCR anidado (nRT-PCR) que se reporta en el Manual de la OIE. La especificidad y la sensibilidad de ambos métodos se evaluaron usando ARN de BTV-4 extraído a partir de diluciones seriadas del virus en medio de cultivo de tejido y en sangre (comenzando a partir de 104.5 TCID50/ml). El límite de detección de cada ensayo en medio de cultivo de tejido fue de 100.5 TCID50/ml y en sangre fue de 101.5 TCID50/ml (rRT-PCR) y 100.5 TCID50/ml (nRT-PCR). Los análisis de curvas de disociación mostraron curvas de amplificación con curvas de disociación específicas (Tm = 84°C ± 0.5°C) y ausencia de amplificación no específica. La sensibilidad diagnóstica se evaluó a partir de muestras clínicas de un ternero bajo condiciones controladas. Estos resultados mostraron que el rRT-PCR, basado en SYBR Green-I, es un ensayo rápido, sensible e igualmente específico en el diagnóstico de animales afectados con BT.
Palabras clave: virus de la lengua azul, RT-PCR anidado, ensayo de RT-PCR en tiempo real basado en SYBR Green-I.
INTRODUCTION
Bluetongue (BT) is an insect-transmitted viral disease of ruminant species, which primarily affects sheep. This disease is caused by the bluetongue virus (BTV) of the genus Orbivirus within the family Reoviridae (1). Twenty-six distinct serotypes of the virus have been identified to date (2, 3, 4).
BT has been listed as a notifiable disease by the World Organization for Animal Health (5). Apart from causing losses through reduced productivity of farm animals, BT infection can also negatively affect the trade of such animals, due to the strict restrictions on the movement of animals and animal products from BT-endemic countries to BT-free countries.
Outbreaks of BT are suspected from clinical symptoms such as fever, depression, excessive drooling, nasal discharges, oral lesions, facial edema, hyperemia of coronary bands, muscle weakness, and reproductive disorders (6).
Virus identification traditionally requires isolation and amplification of the virus in embryonated chicken eggs, tissue culture, or inoculations of susceptible ruminants and the subsequent application of serogroup- and serotype-specific tests. Reverse-transcription polymerase chain reaction (RT- PCR) technology has permitted rapid amplification of BTV RNA in clinical samples, and RT-PCR-based procedures are available. Real-time PCR is allowing development of even more rapid and sensitive tests, and its procedures based in probes for detection of BTV are reported in the OIE Manual (7).
The SYBR Green I-based real-time assay has proven to be one of the most effective tools in the rapid and differential detection of a variety of viral pathogens (8, 9). The current work was aimed to standardize a SYBR Green-I based real-time RT-PCR assay for the detection of bluetongue virus in centinel animals and to compare it with the nested RT-PCR assay reported in the OIE Manual.
MATERIALS AND MÉTHODS
For rRT-PCR assay standardization, the vaccine strain (BTV-4 serotype) used in this study was gently donated by the Central Laboratory of Veterinary, Algete Spain). The virus titer was determined by micro-titration assay using VERO cells cultured in Eagle medium (DMEM) supplemented with L-glutamine, antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin), and 2% fetal calf serum. The TCID50 was calculated using the method of Reed and Muench (10).
A calf without ingesting colostrum, which was fed with milk replacer for the first 4 days and later with milk, was included in this study. It was released to natural conditions under control. The temperature was monitored daily in the morning hours. Blood samples with anticoagulant (3% EDTA) were taken twice a week for twelve days to obtain the fraction of red blood cells in PBS (nRT-PCR and rRT-PCR).
Viral RNA of tissue culture supernatant and whole blood was extracted using a combination of two methods. At first, TRI Reagent® LS (SIGMA, San Louis, Missouri, E.U.A.) was used according to the manufacturer's instructions until the aqueous phase was obtained. Absolute ethanol was added to this phase, and from this step the extraction continued according to the recommendations of the suppliers of QIAamp® Viral RNA Mini Kit (Qiagen®,GmbH). ARN was resuspended in 60 ìl of elution buffer.
First strand complementary DNA (cDNA) was synthesized using Moloney-Murine leukemia virus reverse transcriptase (MMLV-RT) (Promega, Madison, WI, USA) according to manufacturer's instructions.
The presence of BTV was tested by nRT-PCR by using primers targeted to segment 5, encoding the highly conserved non-structural protein NS1, recommended by the OIE (7). The primers used for amplification were Primer A: 5'-GTT-CTC-TAG-TTG-GCA-ACC-ACC-3' and Primer B: 5'-AAG-CCA-GAC-TGT-TTC-CCG-AT-3' as forward and reverse primers, respectively. The nested RT-PCR primers were Primer C: 5'-GCA-GCA-TTT-TGA-GAG-AGC-GA-3' and Primer D: 5'-CCC-GAT-CAT-ACA-TTG-CTT-CCT-3'. The first reaction amplifies a segment of 274 pb, and the second one amplifies a segment of 101 pb.
The rRT-PCR was optimized and carried out using the SYBR Green Quantitative RT-PCR Kit (Roche Diagnostics, Manheim, Germany). Different concentrations of primers (0.4, 0.5, 0.6 mM) and MgCl2 (1.0, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5 mM) were evaluated. All rRT-PCR reactions were performed in duplicate and conducted on the LightCycler 1.5 (Roche Diagnostics, Manheim, Germany) instrument. The following protocol was used: 10 min at 95°C, followed by 35 cycles at 95°C for 15 s, 58°C for 5 s and 72°C for 10 s. After the PCR cycles, a DNA melting curve was generated (0 s at 95°C, 15 s at 68°C, with a ramping time of 20°C/s and 0 s at 95°C with a ramping time of 0.1°C/s) in order to discriminate between specific amplicon and non-specific amplification products.
In this study, the assay detection limit was determined by the efficacy of the entire method, including the sensitivity of the primers and the optimization of the procedure. For the analytical sensitivity evaluated by infectious doses, cell culture-grown virus (104.5 EID50/ml) vaccine BTV-4 strain, was 10-fold serially diluted in MEM medium and RNA from each dilution was extracted. Evaluation of the analytical sensitivity of the method was done by testing each dilution in three replicates.
The matrix effect of blood on the analytical sensitivity of the rRT-PCR assay was assessed by analysing RNA isolated from a sequential 10-fold dilution series of titrated 104.5 EID50/ml of the vaccine BTV-4 strain in negative blood samples obtained from calf. The matrix was spiked at a 9:1 ratio (v:v) with a 10-fold dilution series of vaccine BTV-4 strain. Three replicates per dilution were prepared and tested using LightCycler_Software 4.05.415 (Roche Applied Science, Mannheim, Germany).
The assay specificity was tested on RNA of bovine viral diarrhea (BVD) and DNA of infectious bovine rhinotracheaeitis (HVB-1) and bovine herpes mammillitis (HVB-2). Each strain used was tested in triplicate.
Specimens were considered BTV positive when (i) the Ct value was below 35; (ii) the curve in the amplification plot showed exponential increase, and (iii) a BTV-specific melting curve (matching with those of the positive controls in the same run) was obtained (Fig.1).
The optimization of the rRT-PCR reaction components and alignment temperature was performed. The most suitable combination of the primers concentration and MgCl2 was meticulously selected.
RESULTS AND DISCUSSION
Finally, 0.6 mM and 2.5 mM concentrations of primers and MgCl2 were established and used in further experiments. The final PCR mixture contained 1.2 ml each of forward and reverse primers (final concentration of each, 0.6 mM), 2 ml of Fast Start DNA Master SYBR Green I (2 ml), 1.2 ml of MgCl2 (final concentration 2.5 mM), 2 ml of template, and made up to 18 ml with nuclease free water. The optimal alignment temperature was 58oC.
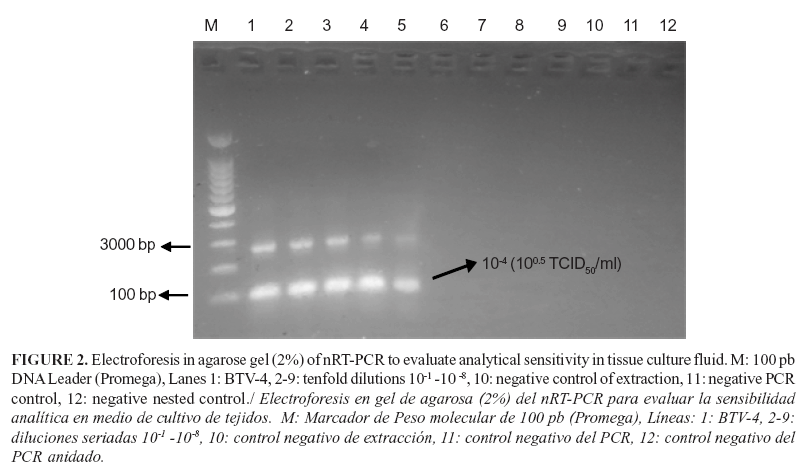
In the evaluation of the analytical sensitivity of BTV-4 RNA extracted from tenfold serially diluted tissue culture medium, the rRT-PCR assay yielded curves of amplification with specific melting curves of 84°C± 0.5°C. The detection limit was 100.5 TCID50/ml (Fig.1A y B). The specific amplifications were also confirmed by the detection of a band of 274 bp by agarose gel electrophoresis (data not shown). Similar results were obtained when the detection limit of nRT-PCR was evaluated. Bands of the expected size (101 bp) were vizualized in electrophoresis in agarose gel when different dilutions of template were tested (Fig. 2). No amplifications were observed in the no template control (NTC) or negative PCR controls.
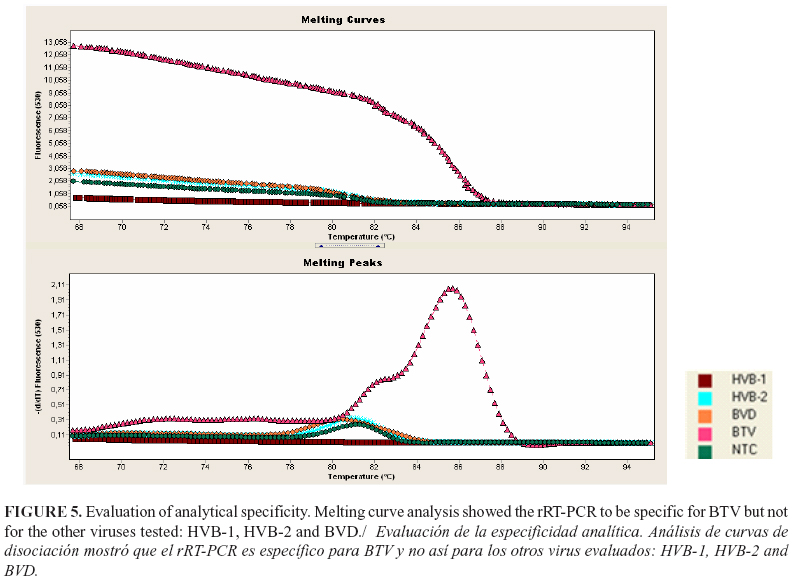
The detection limit in blood was 101.5 TCID50/ml (rRT-PCR) and 100.5 TCID50/ml (nRT-PCR) (Fig. 3 and 4 respectively). The nRT-PCR was able to detect one dilution more than the rRT-PCR. The specificity of the amplification was carried out by melt curve analysis of the amplified products in rRT-PCR. No amplifications curve were obtained with any of the other viruses tested (Fig. 5).
When the rRT-PCR assay for detection of BTV was applied in blood samples from one infected naturally animal, only nine of the twelve samples screened resulted positive, whereas the twelve samples were positive by the nRT-PCR assay. The lack of coincidence in the results between both methods was at the first three evaluation times. These samples were weakly positive by nRT-PCR and amplification bands were only obtained in the second round. It is indicative of a low viral load, reason for which they were not detected in the rRT-PCR. These results agreed with those obtained when the analytic sensibility in blood was evaluated, where the nRT-PCR detected a dilution more than the rRT-PCR. Although the nRT-PCR was more sensitive, the results are comparable.
The current rRT-PCR assay constitutes an attractive and alternative method to the nRT-PCR reported in the Manual of the OIE (7) because of its advantages including not requiring the agar gel electrophoresis to show the amplification of the target sequence, which severely limits the speed of testing. Besides, this assay has an increased sensitivity, improved ease of use, improved throughput and a reduced risk of cross contamination (11).The SYBR Green I RT-PCRs coupled to melting curves analysis provides a reliable method for detection and differentiation of nucleic acid targets (9). In addition, this kind of assay is able to discriminate between different viral strains based on genomic sequence variability (8).
The rRT-PCR assay using NS1-specific primers reported in this study was found to be specific, and a rapid assay in the detection of BTV in blood samples of BTV-affected animals. However, more blood samples from animals during natural outbreaks must be subjected to the rRT-PCR assay developed by us to be able to correlate viral load with disease severity.
REFERENCES
1. Mertens PP, Diprose J, Maan S, Singh KP, Attoui H, Samuel AR. Bluetongue virus replication, molecular and structural biology. Vet Ital. 2004;40:426-437.
2. Chaignat VG, Scherrer W, Hilbe N, Ehrensperger M, Batten F, Corteyn C, et al. Toggenburg orbivirus, a new bluetongue virus: initial detection, first observations in field and experimental infection of goats and sheep. Vet Microbiol. 2009;138:11-19.
3. Maan S, Maan NS, Nomikou K, Batten C, Antony F, Belaganahalli MN, et al. Novel bluetongue virus serotype from Kuwait. Emerg Infect Dis. 2011;17:886-889.
4. Batten CA, Henstock MR, Bin-Tarif A, Steedman HM, Waddington S, et al. Bluetongue virus serotype 26: Infection kinetics and pathogenesis in Dorset Poll sheep. Vet Microbiol. 2012;157:119-124.
5. World Organization for Animal Health (2011) OIE Listed Diseases 2011. Paris: OIE.
6. Maclachlan NJ, Crafford JE, Vernau W, Gardner IA, Goddard A, et al. Experimental reproduction of severe bluetongue in sheep. Vet Pathol. 2008;45:310-315.
7. OIE Terrestrial Manual 2009. Chapter 2.1.3. Bluetongue and Epizootic Haemorrhagic Disease NB: Version adopted by the World Assembly of Delegates of the OIE in May 2009.
8. Varga A, James D. Real-time RT-PCR and SYBR Green I melting curve analysis for the identification of Plum pox virus strains C, EA, and W: Effect of amplicon size, melt rate, and dye translocation. J Virol Methods. 2006;132:146-153.
9. Tam S, Clavijo A, Engelhart EK. Fluorescence-based multiplex real time RT-PCR assays for the detection and serotype determination of foot and mouse disease virus. J Virol Methods. 2009;161:183-191.
10.Reed LJ, Muench H. A simple method of estimating fifty percent end points. Am J Hyg. 1938;27:493-497.
11.Bélak S. Molecular diagnosis of viral diseases, present trends and future aspects a view from the OIE Collaborating Centre for the application of polymerase chain reaction methods for diagnosis of viral diseases in veterinary medicine. Vaccine. 2007;25:5444-5452.
Recibido: 2-2-2016.
Aceptado: 11-3-2016.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}