










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Hematología, Inmunología y Hemoterapia
Print version ISSN 0864-0289
Rev Cubana Hematol Inmunol Hemoter vol.30 no.1 Ciudad de la Habana Jan.-Mar. 2014
ARTÍCULO ORIGINAL
Aspectos diagnósticos, evolutivos y terapéuticos de la leucemia mieloide crónica
Diagnostic, evolutive and therapeutic aspects of chronic myeloid leukemia
Dr. Onel M. Avila-Cabrera, Dra. Yesi C. Expósito-Delgado, Dra. Leslie González-Pinedo, Dr. Edgardo Espinosa-Estrada, Dr. Carlos Hernández-Padrón, Dr. Luis G. Ramón-Rodríguez, Dra. Lissette Izquierdo-Cano, Dr. Gelquin Mustelier-Celza, Dr. Wilfredo Roque-García, DrC. Antonio Bencomo-Hernández
Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
Introducción: la leucemia mieloide crónica (LMC) es un síndrome mieloproliferativo crónico caracterizado por la presencia de una alteración citogenética en las células proliferantes, el cromosoma Filadelfia (Ph), que da lugar a la formación de un gen híbrido BCR-ABL, fundamental en la patogénesis de la enfermedad.
Objetivo: describir el comportamiento de esta enfermedad en los pacientes tratados en el Instituto de Hematología e Inmunología.
Métodos: se estudiaron las características de esta enfermedad en sus aspectos diagnósticos, evolutivos y terapéuticos, en los pacientes atendidos desde marzo de 1974 hasta junio de 2012.
Resultados: el grupo de edad que predominó para ambos sexos fue de 30 a 39 años. El 21 % de los pacientes se encontraban asintomáticos en el momento del diagnóstico. La esplenomegalia fue el signo predominante en el 64 % de los pacientes. Los hallazgos iniciales más significativos del hemograma consistieron en leucocitosis, basofilia y anemia. El 81 % de los casos se encontraba en fase crónica al inicio de la enfermedad. La mayoría de los pacientes debutaron con niveles de LDH elevados. Se observó el cromosoma Filadelfia en el 68 % de los pacientes a quienes se les realizó estudio citogenético. El estudio del reordenamiento del gen BCR/ABL se realizó en el 70 % de los casos, siendo positivo en su totalidad. La media de supervivencia en relación con el tratamiento fue mayor en los pacientes tratados con mesilato de imatinib desde el momento del diagnóstico (11.7 años). La media de supervivencia global es de 11.44 años y la mediana es de 8.18 años.
Conclusiones: todos los parámetros demográficos, clínicos, de laboratorio y terapéuticos coincidieron con lo descrito en la literatura, excepto la edad de aparición de la LMC que evidenció una disminución.
Palabras clave: Leucemia mieloide crónica, supervivencia, imatinib, interferón.
ABSTRACT
Introduction: Chronic myeloid leukemia (CML) is a chronic myeloproliferative syndrome characterized by the presence of a citogenetic alteration in proliferant cells, the Philadelphia chromosome (Ph), giving rise the formation of a hybrid gene BCR-ABL, with a fundamental role in the pathogenesis of the disease.
Objective: To describe the clinical behavior of the illness in patients treated at the Institute of Hematology and Immunology.
Methods: Characteristics according to diagnose, evolution and therapeutic aspects of the patients treated at from March 1974 to June 2012 were studied.
Results: The age group that prevailed for both sexes was 30-39 years; 21 % of the patients were asintomatic at diagnosis. Splenomegaly was the predominant sign in 64 % of the patients. The most significant initial discoveries on blood film were leucocytosis, basophillia and anemia; at debut 81 % of the patients were in chronic phase. Most patients debuted with high levels of LDH. Ph chromosome was observed in 68 % of individuals with cytogenetc studies. The BCR/ABL gene was detected in every patient with molecular studies. The mean of survival in relation to treatment was higher in patients treated with imatinib mesilate at diagnosis (11.7 years). The mean of global survival was 11.44 years with the median of 8.18 years.
Conclusion: All demographic, clinical, laboratory and therapeutic parameters coincided with the literature with the exception of a lower age at onset of CML.
Keywords: Leukemia chronic mieloide, survival, imatinib, interferon.
INTRODUCCIÓN
La leucemia mieloide crónica (LMC) es un síndrome mieloproliferativo crónico que tiene su origen en una célula madre pluripotencial común a las tres series hematopoyéticas. Es uno de los pocos ejemplos de enfermedad maligna donde un único defecto molecular es responsable de la mayoría de los casos.1-3
Está caracterizado por la presencia en la mayoría de los pacientes, de una alteración citogenética en las células proliferantes, el cromosoma Filadelfia (Ph).4-6 Dicha alteración genética refleja la existencia de una translocación recíproca entre los brazos largos de los cromosomas 9 y 22 {t (9;22)(q34;q11)}, que da lugar a la formación de un gen híbrido BCR-ABL, que es fundamental en la patogénesis de la enfermedad.4
El agente etiológico que más claramente se ha relacionado con el desarrollo de la LMC es la exposición a radiaciones ionizantes.3,7 También se ha asociado a la administración de radioterapia para el tratamiento de la espondilitis anquilosante y el cáncer de cuello uterino.8 Los leucemógenos químicos, como el benceno y agentes alquilantes, no se han identificado como agente causante de LMC, con excepción de los agentes inhibidores de la topoisomerasa II del ADN, que tienen propensión a inducir leucemia con t (9;22).9
No existe asociación hereditaria, familiar, geográfica, étnica o económica a la aparición de la LMC.10
La LMC representa del 10 al 15 % del total de leucemias en los individuos adultos y su incidencia se estima en un caso nuevo por 100 000 habitantes por año. En Norteamérica 5 050 casos son diagnosticados cada año 5 y la tasa de mortalidad a causa de ella es de aproximadamente 0,9 por 100 000 personas.3,9
La LMC evoluciona de una manera bi o trifásica. El paso de una fase a otra se define mediante la evolución de parámetros clínicos y analíticos.9,11 La fase inicial, conocida como fase mielocitaria o crónica, tiene escasas manifestaciones clínicas y se puede prolongar durante años, con una mediana de duración de 5 a 6 años. La fase de aceleración con síntomas sistémicos y cambios en la proporción de los elementos inmaduros en la sangre periférica y aparición de alteraciones citogenéticas complejas, dura 6 - 9 meses y la crisis blástica de transformación en leucemia aguda (LA) que dura aproximadamente de 3 - 6 meses.
Desde el punto de vista hematológico y clínico se caracteriza por leucocitosis, trombocitosis y esplenomegalia, aunque algunos pacientes pueden debutar sin síntomas evidentes de la enfermedad. Se señala que alrededor del 50 % de los casos muestran al inicio leucocitosis, a expensas de neutrófilos y mielocitos, con presentación de todos los estados de maduración. También se puede encontrar basofilia absoluta, anemia y eosinofilia.3,11
El tratamiento de la LMC está en evolución a medida que se realizan nuevos ensayos terapéuticos.3,12,13 De igual manera, los objetivos terapéuticos han evolucionado, desde la obtención de una respuesta hematológica (RH), a la de una respuesta molecular (RM), pasando por una respuesta citogenética completa (RCC).14
Por no existir el precedente de investigaciones acerca del comportamiento de la enfermedad en Cuba se decidió estudiar las características de la leucemia mieloide crónica según aspectos diagnósticos, evolutivos y terapéuticos en el Instituto de Hematología e Inmunología (IHI) entre marzo de 1974 y junio de 2012.
MÉTODOS
Se realizó un estudio descriptivo transversal que incluyó 99 pacientes atendidos en el IHI entre marzo de 1974 y junio de 2012, que cumplieron los requisitos de diagnóstico de LMC y edad de 20 años o más.
Se analizaron las historias clínicas de los pacientes con LMC para obtener los síntomas al inicio de la enfermedad y los hallazgos al examen físico. Se obtuvieron, además, las variables hematológicas al inicio (cifras de hemoglobina, conteo de leucocitos y plaquetas) realizados por las técnicas hematológicas habituales15; los parámetros bioquímicos séricos (determinación de la actividad de la LDH, fosfatasa alcalina leucocitaria (FAL)16 y muramidasa sérica17) y los estudios morfológicos y anatomopatológicos de la médula ósea, así como el cariotipo realizado por la técnica de banda G y el reordenamiento del gen BCR-ABL por la reacción en cadena de la polimerasa por reverso transcriptasa.
Los criterios diagnóstico de las fases de la LMC fueron los establecidos por la OMS.18
Se adoptaron los siguientes criterios:
- Criterio diagnóstico de la LMC: detección del cromosoma Ph o el reordenamiento del gen BCR/ABL.
- Anemia: cifras de hemoglobina inferiores a 120 g/L en mujeres y 130 g/L en hombres.
- Leucocitosis ligera: conteo de leucocitos: 11.6 19 x 109/L; moderada: 20 49 x 109/L; severa: mayor o igual de 50 x 109/L.
- Trombocitopenia: conteo de plaquetas inferior a 150 x 109/L.
- Trombocitosis: conteo de plaquetas superior a 450 x 109/L.
- Eosinofilia: por encima de 0,50 x 109/L.
- Basofilia: por encima de 0,150 x 109/L.
El procesamiento estadístico se realizó con el programa SPSS versión 19. Se utilizaron procederes estadísticos univariados, se determinó la media y desviación estándar en variables cuantitativas y distribuciones de frecuencia absolutas y relativas con porcentajes en las variables cualitativas. Se realizó el análisis de supervivencia con el método no paramétrico de Kaplan Meier con todos los pacientes y por tratamientos.
RESULTADOS
De los 99 pacientes incluidos en el estudio, el 52 % era del sexo masculino y el 48 % del sexo femenino. El grupo de edad predominante en los pacientes de sexo masculino fue de 30 a 39 años que representó el 27 %. En los pacientes del sexo femenino fueron más frecuentes los grupos de edades entre 30 y 49 años (29 %).
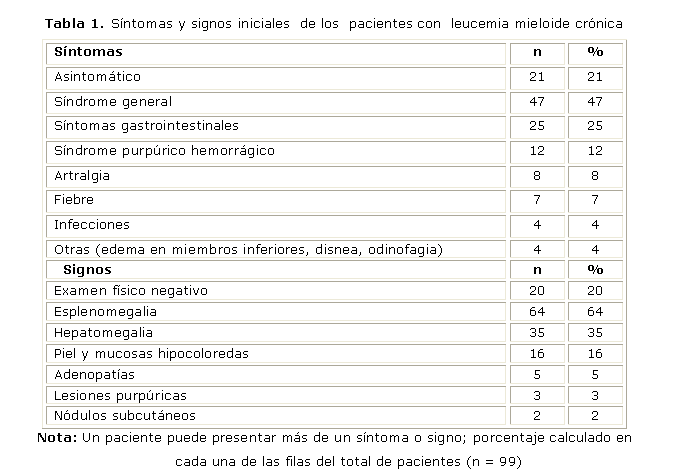
El 21 % de los pacientes estudiados se encontraba asintomático en el momento del diagnóstico. Los síntomas iniciales más frecuentes fueron: síndrome general (47 %), seguido de síntomas gastrointestinales que representaron el 25 % (tabla 1).
En el 20 % de los pacientes con LMC no se encontraron datos positivos al examen físico. La esplenomegalia fue el signo predominante en el 64 % de los pacientes y seguidamente, la hepatomegalia (35 %) (tabla 1).
Los hallazgos iniciales del hemograma en todos los pacientes consistieron en leucocitosis (86 %), basofilia (78 %) y anemia (74 %). También se encontró mielemia en el 41 % de los enfermos, trombocitosis en el 31 % y eosinofilia en el 30 % (tabla 2).
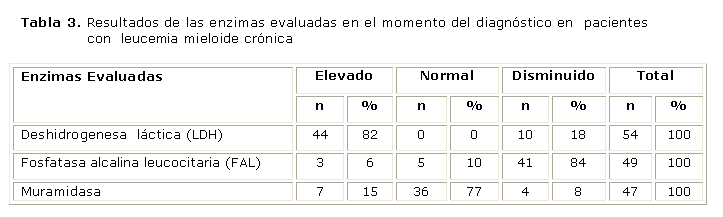
La mayoría de los pacientes debutó con niveles de LDH elevados, FAL disminuida y muramidasa con valores dentro del rango normal (tabla 3).
El 81 % de los casos se encontraba en fase crónica al inicio, el 14 % en crisis blástica y el 5 % en fase acelerada.
En el 69 % del total de la muestra, se observó el cromosoma Ph en el estudio citogenética, y reordenamiento positivo del gen BCR/ABL en el 100 %; la positividad de ambos estudios fue del 84 %.
La media de supervivencia en relación con el tratamiento fue de 7,4 años para los pacientes que recibieron tratamiento con busulfan con interferón alfa (IFN-a) o sin este; de 8,3 años para los que recibieron interferón de primera línea y mesilato de imatinib como segunda línea de tratamiento; y de 11,7 años para aquellos que fueron tratados con mesilato de imatinib desde el momento del diagnóstico (figura 1).
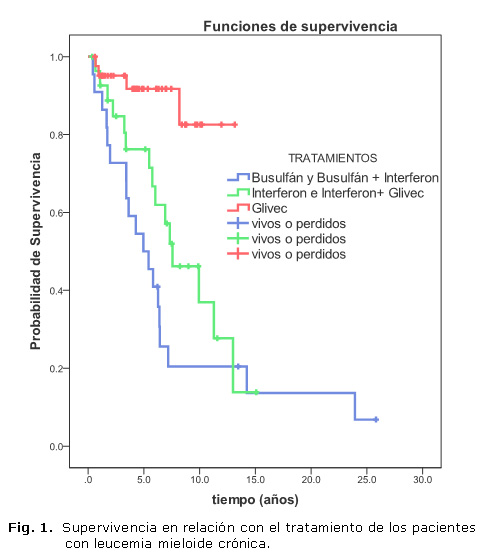
La media de supervivencia global fue de 11,44 años, con un intervalo de 8,6 a 14,2 años y una mediana de 8,18 años (figura 2).

DISCUSIÓN
En la serie estudiada se observó un ligero predominio del sexo masculino. Este hallazgo coincide con lo encontrado en la literatura que plantea que existe un discreto predominio de varones con una relación hombre:mujer de 1.4:1. 9
Predominaron los grupos de edades entre 20 y 49 años en el sexo femenino y entre 30 y 39 años en el sexo masculino, ligeramente inferior a la mediana de edad de aparición que plantean otros autores que es entre 50 y 65 años, aunque el 10 % de los casos debutan entre los 5 y 20 años.10
El 20 % de los pacientes se encontraban asintomáticos al inicio de la enfermedad. En una proporción frecuente de enfermos la dolencia se descubre casualmente cuando se hacen recuentos de células sanguíneas en un estudio médico rutinario.19 De acuerdo con la literatura, entre el 10 - 30 % de los pacientes son asintomáticos al debut de la enfermedad.20
Los síntomas encontrados coinciden con los comunicados en la literatura como más frecuentes, con predominio del síndrome general y los síntomas gastrointestinales.21 En la exploración física, la esplenomegalia fue el hallazgo característico, similar a lo comunicado por otros investigadores. La esplenomegalia, presente en aproximadamente el 90 % de los pacientes, está disminuyendo su frecuencia en el momento del diagnóstico al hacerse este más precozmente en la actualidad. El segundo hallazgo más frecuente fue la hepatomegalia.3
El diagnóstico de LMC es frecuentemente precedido por un período de varios meses en que los pacientes presentan síntomas generales atribuibles a un estado de hipermetabolismo provocado por el aumento del recambio granulocítico, o bien a molestias por la esplenomegalia. Otras manifestaciones clínicas menos frecuentes son los dolores óseos, hemorragias, crisis de gota, litiasis renal, priapismo o síntomas de leucoestasis por hiperleucocitosis.20
Un alto porcentaje de los pacientes con LMC presentan al inicio recuento total de leucocitos elevado por encima de 25 x 109/L; la mitad de los pacientes tienen recuentos de leucocitos por encima de 100 x 109/L y el recuento global aumenta progresivamente en los enfermos no tratados. Hay células de todos los estadios madurativos de la granulopoyesis, con predominio de las formas más maduras, salvo por la mayor proporción de mielocitos que de metamielocitos, y generalmente de aspecto normal; se detectan también basofilia y eosinofilia. Por lo general, el porcentaje inicial de blastos en la sangre periférica es bajo (menos del 3 %).
El recuento de plaquetas está elevado en el 40 % de los pacientes al momento del diagnóstico y es normal en el resto; no son inusuales los recuentos de plaquetas mayores de 1 000 x 109/L. Ocasionalmente el recuento de plaquetas puede estar por debajo de la normalidad en el momento del diagnóstico, pero esto habitualmente indica la progresión inminente a la fase acelerada de la enfermedad. Estos hallazgos; comunicados por la mayoría de los autores coinciden con los encontrados en la serie analizada, excepto para el recuento de plaquetas donde la mayoría de los pacientes estudiados presentaron valores dentro de parámetros normales, lo que podría deberse al número de casos analizados en el estudio.
Se encontraron niveles elevados de la LDH en la mayoría de los pacientes, lo que coincide con los hallazgos de otros autores. La actividad de la FAL estaba baja o ausente en más del 90 % de los pacientes con LMC. El ARNm de la FAL es indetectable en los neutrófilos de los pacientes con LMC; la actividad aumenta hacia la normalidad en presencia de inflamación o infecciones, cuando el recuento total de leucocitos disminuye, y con el tratamiento. Las elevaciones de los niveles séricos y urinarios de lisozima son características de la leucemia con mayores componentes monocíticos y no son rasgos de LMC. 20-22 Todo lo anterior explica también la presencia de FAL disminuida y muramidasa normal en la mayoría de los pacientes.
Este estudio se comportó de forma similar a lo descrito en la literatura, donde el 80 % de los pacientes se diagnostica en fase crónica, el 20 % restante evoluciona directamente a crisis blástica sin previo paso por fase acelerada. De estos, aproximadamente el 5 % se presenta directamente en fase acelerada o en crisis blástica.20
El estudio citogenético de la médula ósea reveló la presencia del cromosoma Ph en la mayoría de los pacientes estudiados. Las investigaciones demuestran presencia del cromosoma Ph clásico en el 70 % de los pacientes en la fase crónica de la enfermedad. La médula y las células sanguíneas nucleadas de más del 90 % de los pacientes con signos clínicos y de laboratorio con criterios para el diagnóstico de LMC, contienen el cromosoma Ph, el que está presente en todas las estirpes de células sanguíneas (eritroblastos, granulocitos, monocitos, megacariocitos, progenitores de linfocitos T y B)3. La translocación (9;22) no es exclusiva de la LMC (95 %), también puede observarse en el 5 % de las leucemias linfoides agudas del niño, entre el 15 - 30 % del adulto, en el 2 % de todas las leucemias mieloides agudas y en el 20 - 30 % de adultos sanos, atribuible a la inestabilidad del genoma.23
El reordenamiento del gen BCR/ABL fue positivo en la totalidad de los pacientes en que se realizó. Las técnicas de análisis molecular demuestran reordenamiento BCR/ABL en todos los casos de LMC Ph-positiva y en un tercio de los casos de LMC Ph-negativa.24 En una pequeña proporción de pacientes con una enfermedad clínica análoga a la LMC, los estudios citogenéticos no revelan un cromosoma clásico, variante o enmascarado. La capacidad para identificar las consecuencias moleculares de la t(9;22) está en el reordenamiento del BCR, los transcriptos de ARNm del gen de fusión mutante y la proteína p210, que son pruebas diagnósticas complementarias al análisis citogenético.25,26
De los esquemas de tratamiento utilizados en los pacientes de esta serie, el busulfan fue con el que se logró una supervivencia menor, aunque en la mayoría de los casos recibieron también IFN-a. El busulfán fue el tratamiento fundamental para la LMC en fase crónica hasta que se incorporaron la hidroxiurea y IFN-a. El objetivo de este tratamiento era controlar la fase crónica de la enfermedad tratando de reducir en lo posible la morbimortalidad. De esta forma se obtenía el descenso de las cifras de leucocitos circulantes, lo que influía favorablemente en las secuelas inmediatas de la leucocitosis excesiva, la reducción de la esplenomegalia y el aumento del hematocrito, así como la mejoría subjetiva del estado general. Estas respuestas no eran muy duraderas; salvo en casos muy esporádicos, se conseguía disminuir el porcentaje de cromosoma Ph.27
En la década del 80 se demostró que el IFN-a (natural o recombinante) era más efectivo y menos tóxico que la hidroxiurea. Los pacientes que recibían este tratamiento mostraban una recuperación satisfactoria de los síntomas de la enfermedad, por lo que a partir de entonces se empleó en la mayoría de los pacientes con LMC como tratamiento único o en combinación con otro citostático; la citosina arabinósido (Ara-C). De esta forma, la supervivencia de los pacientes tratados exclusivamente con este fármaco fue ligeramente superior.28
La media de supervivencia superior se encontró en los pacientes tratados con mesilato de imatinib, lo que está demostrado en diferentes estudios. La supervivencia global mostrada en esta serie está influenciada por los pacientes tratados con esta droga. El tratamiento con mesilato de imatinib logra una respuesta hematológica, clínica y molecular superior a la alcanzada con el IFN- recombinante. Esta terapia se está empleando desde finales de la década del 90 en pacientes de nuevo diagnóstico y en aquellos que han presentado resistencia o intolerancia al INF-a recombinante.29,30
Según las últimas recomendaciones del panel de expertos de la Red Europea de Leucemia, el tratamiento con imatinib debe ser la primera opción terapéutica y la respuesta a este el parámetro a considerar para la indicación de alotrasplante.31,32
Recientemente se ha planteado la reincorporación del IFN-a en el tratamiento de la LMC, apoyado por las investigaciones realizadas sobre los mecanismos de acción de este fármaco que sugieren que puede tener un efecto citotóxico directo sobre las células malignas, promover la inducción de respuestas inmunes antitumorales y de genes proapoptósicos, inhibir la angiogénesis y promover la introducción en el ciclo celular proliferativo de células madre malignas que se mantenían en estado de reposo.33 Se ha publicado recientemente sobre las ventajas de su uso combinado con los inhibidores de la actividad tirosina cinasa, fundamentalmente en los pacientes que presentan la mutación T315I (sustitución de tirosina por isoleucina en el aminoácido 315), que induce resistencia no solo al imatinib sino también a inhibidores de segunda generación, como el nilotinib y el dasatinib.34
Sin embargo, debemos recordar que, a la espera de un mayor seguimiento de los pacientes tratados con nuevos inhibidores de la actividad tirosina cinasa, el alotrasplante es el único procedimiento que, hasta el momento, ha demostrado potencial curativo definitivo.9,35
REFERENCIAS BIBLIOGRÁFICAS
1. Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999, Apr 29; 340: 1330-40.
2. Chakraborty S, Stark JM, Sun CL, Modi H, Chen W, O'Connor TR, et al. Chronic myelogenous leukemia stem and progenitor cells demonstrate chromosomal instability related to repeated breakage-fusion-bridge cycles mediated by increased nonhomologous end joining. Blood. 2012 Jun 28; 119(26):6187-97.
3. Liesveld JL, Lichtman MA. Leucemia mielógena crónica y trastornos relacionados. In: Beutler E, Lichtman MA, Coller BS, editors. Williams Hematología. 6ta ed. Madrid: Marbán Libros; 2007. p. 815-38.
4. Faderl S, Talpaz M, Estrov Z, O´Brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia N Engl J Med 1999 Jul; 341(3): 164-72.
5. Goldman JM. Chronic Myeloid Leukemia - Past, Present, and Future. Semin Hematol. 2003 Jan; 40(1):1-3.
6. Marcucci G, Perrotti D, Caligiuri MA. Understanding the molecular basis of imatinib mesylate therapy in chronic myelogenous leukemia and the related mechanisms of resistance. Commentary re: A. N. Mohamed et al., The effect of imatinib mesylate on patients with Philadelphia chromosome-positive chronic myeloid leukemia with secondary chromosomal aberrations. Clin. Cancer Res, 9: 1333-1337, 2003. Clin Cancer Res. 2003 Apr; 9(4):1248-52.
7. Geary CG. Historical review. The story of chronic myeloid leukaemia. Br J Haematol. 2000; 110(3):2-11.
8. Goldman JM, Melo JV. Chronic Myeloid Leukemia - Advances in Biology and New Approaches to Treatment. N Engl J Med. 2003; 349:1451-64.
9. García-Gutiérrez JV. Factores pronósticos en leucemia mieloide crónica Filadelfia positiva en pacientes tratados con inhibidores de la tirosincinasa [Tesis doctoral]. Granada: Universidad de Granada; 2008.
10. Quintás-Cardama A, Cortes JE. Chronic Myeloid Leukemia: Diagnosis and Treatment. Mayo Clin Proc. 2006; 81(7):973-88.
11. Melo JV, Deininger MW. Biology of chronic myelogenous leukemia—signaling pathways of initiation and transformation. Hematol Oncol Clin North Am. 2004 Jun; 18(3):545-68.
12. Quintás-Cardama A, Cortes JE. Chronic Myeloid Leukemia: Diagnosis and Treatment. Mayo Clin Proc. 2006; 81(7):973-88.
13. Melo JV, Hughes TP, Apperley JF. Chronic Myeloid Leukemia. Hematology Am Soc Hematol Educ Program. 2003:132-52.
14. Aguayo A, Couban S. State-of-the-art in the management of chronic myelogenous leukemia in the era of the tyrosine kinase inhibitors: evolutionary trends in diagnosis, monitoring and treatment. Leuk Lymphoma. 2009 Dec;50 Suppl 2:1-8.
15. Vives Ll, Aguilar J. Métodos de recuento de las células sanguíneas. En: Vives Ll, Aguilar J. Manual de técnicas de laboratorio en hematología. 3ed. Barcelona: Elsevier-Masson; 2006.p.63-140.
16. Diagnosticadores para química clínica y microbiología. En: Helfa Diagnósticos. La Habana: EPB Carlos J. Finlay. Quimera; 2003. [consultado: 12 de junio de 2013]. Disponible en: http://www.biofinlay.sld.cu
17. Parry RM Jr, Chandan RC, Shahani KM. A rapid and sensitive assay of muramidase. Proc Soc Exp Biol Med 1965; 119:384-6.
18. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon: IARC Press; 2008.
19. Besses C, Hernández-Nieto L. Síndromes mieloproliferativos crónicos. Haematologica (esp). 2009; 94(Supl 1):379-96.
20. Cervantes Requena F, Besses Raehel C. Síndromes mieloproliferativos crónicos. En: Farreras P, Rozman C. ed. Medicina Interna. Madrid: Elsevier España SA; 2010. p. 1715-27.
21. Greer JP, Foerster J, Lukens JN, Rodgers GM, Parackevas F, Glader B ed. Wintrobe´s Clinical Hematology. 12 ed. Philadelphia: Lippincott Williams & Wilkins. 2009.
22. Cervantes Requena F. Leucemia mieloide crónica. En: García Conde J, San Miguel JF, Sierra J, Urbano Ispizua A, Vicente V, Vives Corrons JL. ed. Hematología. Madrid: Aran Ediciones SL; 2003.p.959-69.
23. Jones D, Chen SS, Jabbour E, Rios MB, Kantarjian H, Cortes J. Uncommon BCR-ABL kinase domain mutations in kinase inhibitor-resistant chronic myelogenous leukemia and Ph+ acute lymphoblastic leukemia show high rates of regression, suggesting weak selective effects. Blood. 2010 Jul 1; 115(26):5428-9.
24. Norkin M, Schiffer CA. Molecular monitoring of BCR-ABL transcripts in patients with chronic myelogenous leukemia: is high sensitivity of clinical value? Curr Hematol Malig Rep. 2010 Apr; 5(2):88-94.
25. Fugazza G, Garuti A, Marchelli S, Miglino M, Bruzzone R, Gatti AM, et al. Masked Philadelphia chromosome due to atypical BCR/ABL localization on the 9q34 band and duplication of the der(9) in a case of chronic myelogenous leukemia. Cancer Genet Cytogenet. 2005 Dec; 163(2):173-5.
26. Liu XP, Liu SH, Li CW, Bo LJ, Qin S, Dai Y, et al. Analysis and identification of variant Ph chromosome translocation in patients with chronic myelogenous leukemia by conventional cytogenetics and fluorescence in situ hybridization. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2004 Jun; 12(3):298-303. (Artículo en chino)
27. Santos FP, Kantarjian H, Quintas-Cardama A, Cortes J. Evolution of therapies for chronic myelogenous leukemia. Cancer J. 2012 Nov-Dec;17(6):465-76.
28. Garcia-Manero G, Talpaz M, Giles FJ, Cortes J, Faderl S, O'Brien S, et al. Treatment of Philadelphia chromosome-positive chronic myelogenous leukemia with weekly polyethylene glycol formulation of interferon-alpha-2b and low-dose cytosine arabinoside. Cancer. 2003 Jun 15;97(12):3010-6.
29. Kantarjian H, O'Brien S, Cortes J, Shan J, Giles F, Garcia-Manero G, et al. Analysis of the impact of imatinib mesylate therapy on the prognosis of patients with Philadelphia chromosome-positive chronic myelogenous leukemia treated with interferon-alpha regimens for early chronic phase. Cancer. 2003 Oct 1;98(7):1430-7.
30. Huges TP, Kaeda J, Brandford S, Rudzki Z, Hochhaus A, Hensley ML, et al. Frequency of major molecular responses to imatinib or interferon alpha plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 2003; 349:1423-32.
31. Chronic myelogenous leukemia. Clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2003 Oct; 1(4):482-500.
32. Baccarani M, Dreyling M. Chronic myelogenous leukemia: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009 May; 20 Suppl 4:105-7. doi:10.1093/annonc/mdp143.
33. Kiladjian JJ, Mesa RA, Hoffman R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood. 2011 May 5;117(18):4706-15.
34. Talpaz M, Hehlmann R, Quintás-Cardama A, Mercer J, Cortes J. Re-emergence of interferon-á in the treatment of chronic myeloid leukemia. Leukemia. 2013 Apr;27(4):803-12.
35. Ballestrero A, Cirmena G, Dominietto A, Garuti A, Rocco I, Cea M, et al. Peripheral blood vs. bone marrow for molecular monitoring of BCR-ABL1 levels in chronic myelogenous leukemia, a retrospective analysis in allogeneic bone marrow recipients. Int J Lab Hematol. 2010 Aug; 32(4):387-91.
Recibido: Abril 25, 2013
Aceptado: Julio 29, 2013
Dr. Onel M Avila Cabrera. Instituto de Hematología e Inmunología. Apartado 8070, La Habana, CP 10800, CUBA. Tel (537) 643 8695, 8268. Fax (537) 644 2334. Correo electrónico: rchematologia@infomed.sld.cu

{kind=link}
{kind=link}
{kind=link}