










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introducción
El factor VII (FVII), también llamado proconvertina, es un factor de la coagulación dependiente de vitamina K sintetizado por el hígado a partir del gen ubicado en el cromosoma 13q34.1,2 En comparación a otros factores que participan en las vías de coagulación, su tiempo de vida media es corto (4 - 6 horas).2,3 La deficiencia del FVII se define con la obtención de un valor menor del 70 % de este en plasma. Las personas con los valores menores a 30 % tienen mayor probabilidad de presentar manifestaciones clínicas.2
Existen 2 tipos de déficit del FVII, el congénito y el adquirido.2 La deficiencia congénita tiene una prevalencia estimada de 1/500 000 personas, siendo ésta la más común en cuanto deficiencias congénitas de factores de coagulación.2,4 La deficiencia adquirida puede ser aislada (FVII como único factor de coagulación disminuido) o combinada (con déficit de otros factores de coagulación).2 La forma congénita es bien conocida, pero se conoce poco de la forma adquirida y son pocos los casos reportados en la literatura.5,6
Se presenta el caso de una mujer peruana de 82 años con episodios recidivantes de hemorragia digestiva baja (HDB), tiempo de protrombina (TP) prolongado y déficit aislado del FVII.
Presentación del caso
Paciente femenina de 82 años de edad, natural y procedente del departamento de Lima, con antecedente de hipertensión arterial (HTA) y diabetes mellitus (DM), ambas controladas con enalapril y metformina, respectivamente. Refiere haber tenido ciclos menstruales regulares y no presenta antecedentes familiares de importancia. Señala antecedente quirúrgico (hace más de 20 años) por una hernia umbilical y prolapso vaginal, sin complicaciones.
Ingresó al servicio de urgencias por presentar cuadro clínico de deposiciones semisólidas con sangrado rojo rutilante (aproximadamente 240 mL) de 3 horas de evolución. Al examen físico se evidenció funciones vitales estables y tacto rectal con signos de sangrado reciente. El hemograma no evidenció alteraciones (leucocitos 7 650 células/mm3, hemoglobina 12,7 g/dL y plaquetas 191 000 cel/mm3). Perfil hepático, renal y electrolitos en rango normal. El perfil de coagulación presentó alteraciones con TP prolongado a 40 segundos y aumento del valor del ratio internacional normalizado (INR, por sus siglas en inglés) en 3,2 (Tabla).
Tabla Análisis de laboratorio
| Análisis | Resultado | Valores normales** |
|---|---|---|
| Hemograma | ||
| Hemoglobina (g) | 12.7 | (12-14) |
| Hematocrito (%) | 36,7 | (39-48) |
| VCM (fl) | 90 | (80-99) |
| HCM (pg) | 31 | (33-37) |
| Plaquetas (mm3) | 191 000 | (150 000 - 450 000) |
| Leucocitos (mm3) | 7 650 | (5 000-10 000) |
| Neutrófilos (%) | 56 | (55-75) |
| Perfil de coagulación | ||
| Fibrinógeno (mg/dl) | 3.62 | (2.0-4.2) |
| TP (segundos) | 40 | (9-13) |
| Corrección con plasma* | 25 | (9-13) |
| TTPa (segundos) | 31 | (27-41) |
| INR | 3.52 | (0.90-1.30) |
| Perfil bioquímico | ||
| Fosfatasa Alcalina (U/L) | 79 | (38-126) |
| TGP (U/L) | 30 | (13-69) |
| TGO (U/L) | 52 | (15-56) |
| Bilirrubina total (mg/dl) | 0.3 | (0.2-1.3) |
| Creatinina (mg/dl) | 0.8 | (0.5-0.8) |
| Glucosa (mg/dl) | 119 | (74-106) |
| Calcio total (mg/dl) | 9 | (8.3-10.6) |
| Factores de coagulación | ||
| Factor II (%) | 80 | 60-100 |
| Factor V (%) | 90 | 50-150 |
| Factor VII (%) | 4 | 70-120 |
| Factor IX (%) | 118 | 70-120 |
| Factor X (%) | 99 | 70-120 |
| Perfil autoinmune | ||
| Anticoagulante lúpico | 1 | <1.3 |
| IgM anticardiolipina (MPL-U/ml) | < 7 | < 7 |
| IgG anticardiolipina (GPL-U/ml) | < 10 | < 10 |
| IgG Anti-Beta 2 glicoproteina I (U/ml) | < 8 | < 8 |
| Marcadores tumorales | ||
| CA 125 (UI/ml) | 8.9 | (<30.2) |
| AFP (ng/ml) | 2.6 | (<8.1) |
| CEA (ng/ml) | 1.29 | (0-5) |
VCM: Volumen corpuscular medio; HCM: Hemoglobina corpuscular media; TP: Tiempo de protrombina; TTPa: Tiempo de tromboplastina parcial activado; INR: Internacional normalized ratio; TGP: Transaminasa glutámico pirúvica; TGO: Transaminasa glutámico oxalacética; CA 125: Cancer antigen 125; AFP: Alfafetoproteína; CEA: Carcinoembryonic antigen.
* Mezcla 1:1 con plasma normal
** Según laboratorio del Hospital Nacional “Guillermo Almenara”
Con estos resultados se establece el contexto de HDB y una coagulopatía, por lo que se le transfundió 600 mL de plasma fresco congelado (PFC) (10 mL/kg) y vitamina K por vía endovenosa (EV). Consecuente a ello, la paciente presentó corrección parcial del TP y una resolución completa del cuadro clínico. La falta de corrección completa del TP indujo la sospecha de déficit de factores dependientes de vitamina K, por lo que se solicitó el nivel de actividad de los factores II, V, VII, IX y X. Todos, excepto el FVII (disminuido a 4 %), evidenciaron un rango normal. Como diagnóstico diferencial se planteó la presencia de patología colorrectal, por lo que se programó a la paciente para una colonoscopía. Previo al procedimiento, se administró 2mg de FVII recombinante activado (30 ug/kg) EV en bolo. La colonoscopía evidenció múltiples divertículos en el colon sin sangrado activo.
La evolución de la paciente fue favorable durante los 5 días de su hospitalización, sin evidencia de nuevo sangrado activo. Por ello, fue dada de alta con el diagnóstico de HDB en el contexto de déficit aislado del FVII y se le brindó indicación de manejo por consulta externa. Sin embargo, la paciente no acudió a su consulta externa programada.
Luego de 8 meses del alta hospitalaria, la paciente reingresó al servicio de urgencias por cuadro clínico similar al anterior: HDB de 2 horas de evolución sin repercusión hemodinámica y prolongación del TP. Recibió 600 mL de PFC, hubo remisión de la sintomatología y fue dada de alta con manejo ambulatorio.
En la consulta externa se ampliaron estudios de búsqueda de etiología del déficit de FVII. No se detectaron inhibidores por el ensayo Bethesda, anticoagulante lúpico 1, marcadores tumorales, como CA125, AF y CEA, todos normales (Tabla). En la tomografía corporal total se confirmó ausencia de neoplasia oculta. Se realizó a la hermana estudios de perfil de coagulación y dosaje de FVII, los cuales resultaron estar en rangos normales.
A 18 meses del diagnóstico, la paciente persiste con niveles bajos del FVII, pero asintomática, sin tratamiento, y en seguimiento ambulatorio por hematología y gastroenterología. En la figura se grafica el orden cronológico de sucesos de salud-enfermedad acontecidos con la paciente.
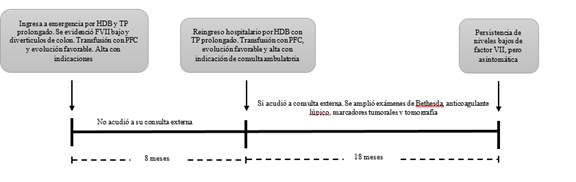
Discusión
La deficiencia del FVII es una enfermedad hemorrágica rara descrita por primera vez en 1951.7 Su forma hereditaria es una enfermedad autosómica recesiva producida por ausencia o déficit de este factor de coagulación.7,8 Se reporta en partes del mundo en donde la consanguinidad es más frecuente.2,7
Las manifestaciones clínicas descritas son menorragia, epistaxis, sangrado de encías, hematomas faciales, hemorragia digestiva, hemartrosis, hematuria y hemorragia intracraneal,8 aunque también pueden ser asintomáticos.2
En el caso de la paciente reportada, la única manifestación que presentó, y por la que acudió, fue la HDB. No fue posible descartar la posibilidad de origen hereditario del déficit del FVII; esto debido a que no fue viable la realización de los estudios genéticos a ella y a sus padres (ya fallecidos). Sin embargo, la ausencia de clínica hemorrágica durante su menstruación o antecedente quirúrgico, más los resultados normales de la prueba hematológica realizada a la hermana, alejan esta posible etiología hereditaria.
La literatura refiere que la forma adquirida del déficit del FVII puede estar combinada con el déficit de algún otro factor de coagulación: déficit desencadenado por mecanismos patológicos como la deficiencia de vitamina K (producida por enfermedades hepáticas) o malabsorción, terapia anticoagulante con antagonistas de la vitamina K, hiperfibrinolisis o coagulación intravascular diseminada.2
En situaciones donde el único factor de coagulación afectado sea el FVII, este déficit se considera como uno aislado; consecuente a ello, se opta por buscar neoplasias, presencia de anticuerpos o inhibidores, trauma mayor, sepsis, anemia aplásica o trasplante de médula ósea.2,9,10,11,12 Una deficiencia aislada adquirida de FVII es una coagulopatía rara; sin embargo, varios autores coinciden que la incidencia podría estar subestimada, esto debido a que la deficiencia aislada adquirida de FVII sin actividad inhibidora no suele considerarse en la práctica habitual.2,6
En relación al caso reportado, este no presentó signos de hepatopatía, carecía de tratamiento anticoagulante con algún antagonista de la vitamina K, los resultados de laboratorio evidenciaron al resto de factores de coagulación dentro de rangos normales (Tabla), en el examen tomográfico no se evidenció neoplasias y no se detectaron anticuerpos o inhibidores; por lo tanto, si bien se trata de un caso aislado de déficit de FVII, no fue posible encontrar algún sustento que explique el déficit único de este factor. Por ello, se consideró diagnosticar a la paciente con el padecimiento de un déficit aislado del FVII con sangrado digestivo como única manifestación y en donde el estudio hematológico de la hermana aleja la causa hereditaria.
La sospecha del déficit de FVII se inició con el resultado de laboratorio de la prolongación del TP; se buscó corregir este TP mediante la corrección con plasma normal (prueba de mezcla) (Tabla). La corrección parcial del TP conllevó a la necesidad de descartar deficiencia asociada a otros factores o por presencia de inhibidores.2 Si bien la paciente presentó evolución clínica favorable tras la administración de hemoderivado, se tiene en consideración que la mayoría los sangrados digestivos bajos se autolimitan en personas sin coagulopatías;13,14 por lo tanto, es difícil afirmar que su mejoría clínica se debió exclusivamente al uso del PFC. También, su forma de diagnóstico es poco usual debido que es frecuente diagnosticar este déficit en estudios preoperatorios.15
En Perú, el acceso al FVII recombinante activado es muy limitado. A pesar de ser la terapia de reemplazo ideal en pacientes con deficiencia del FVII,16 los costos y el trámite administrativo para su obtención dificultan su uso. Por lo tanto, en situaciones de emergencias de pacientes con sangrado y TP prolongado, el uso de PFC es de primera elección.
Se presenta un caso raro de déficit aislado de FVII, en el contexto de una HDB, en una mujer peruana que fue tratada con PFC y evolucionó satisfactoriamente. En la actualidad, la paciente se encuentra estable y acudiendo a sus controles por consulta externa.

