










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
On-line version ISSN 1561-3011
Rev Cubana Invest Bioméd vol.26 no.1 Ciudad de la Habana Jan.-Mar. 2007
Universidad de Valparaíso, Chile
Evaluación de las características biofarmacéuticas y de estabilidad de preparados galénicos sólidos
Dr. Alexis Aceituno, Lic. Karen Navarro y Lic. Cristina Luco
Resumen
Se evaluaron algunas características biofarmacéuticas como disolución y uniformidad de contenido de formas galénicas de tres principios activos lábiles y se evaluó también su estabilidad en condiciones de estrés térmico y humedad relativa en las diferentes formas de envases utilizados. Para satisfacer las necesidades de dosis pediátricas o paciente-específicas que no estén disponibles como productos comerciales, una práctica común de hospital es elaborar y fraccionar dosis en la forma de preparados galénicos. La farmacia asistencial del Hospital Dr. Gustavo Fricke de Viña del Mar, Chile, decidió recientemente, reemplazar el uso de papelillos por el de cápsulas de gelatina dura como el método de fraccionamiento. Sin embargo, existe una carencia de base experimental que permita establecer si el producto conserva su calidad biofarmacéutica y su estabilidad en la forma farmacéutica y el sistema final de envase empleado. Los resultados de este estudio validan el uso de la metodología de elaboración empleada en términos de calidad biofarmacéutica y de estabilidad extrapolada a temperatura ambiente. Estadísticamente no se halló una relación entre la estabilidad y los sistemas de envase para dispensación empleados.
Palabras clave: Calidad biofarmacéutica, estabilidad, cápsulas duras, dosis fraccionadas.
La elaboración de preparados galénicos es una práctica que se hace cada vez más habitual, por la necesidad de contar con dosis no disponibles en las formas farmacéuticas comercialmente utilizables. Dicha necesidad es común por ejemplo en pacientes pediátricos o para el tratamiento específico de pacientes adultos.1 Siguiendo este objetivo, el Hospital Dr. Gustavo Fricke de Viña del Mar, elabora presentaciones farmacéuticas fraccionadas destinadas tanto a pacientes ambulatorios como hospitalizados, con materias primas tanto activas como inactivas obtenidas a granel. Muchas de estas presentaciones específicas se fabrican en pequeños lotes, los cuales se almacenan por períodos autolimitados en dependencias de la Unidad de Farmacia. El período de almacenamiento se estima sobre una base substancialmente empírica, basado en antecedentes del tipo biblográfico.2 Como tal, el período de vida útil podría ser subestimado, sobre todo en el caso de preparados sólidos, para los cuales los cambios fisicoquímicos que de manera eventual podrían afectar la estabilidad de la preparación ocurren quizá en forma mucho más lenta.3,4
El objetivo del presente estudio fue realizar controles biofarmacéuticos y estudiar las características de estabilidad a preparados galénicos sólidos seleccionados, para tener una primera impresión de las particularidades de su calidad, utilizando un protocolo experimental sencillo que de manera eventual podría extenderse en su aplicación a otros tipos de preparados galénicos.
Métodos
Los preparados galénicos seleccionados fueron cápsulas de gelatina dura de 3 principios activos de reconocida inestabilidad5-7 (Furosemida, Ranitidina clorhidrato y Codeína fosfato) y, además, corresponden al volumen mayor de producción de la unidad galénica del hospital. Con la finalidad de detectar potenciales cambios de la calidad biofarmacéutica o de estabilidad en función de su potencia, se estudió el comportamiento de 2 dosis para cada uno de los fármacos: la mayor y la menor dosis dispensada. El análisis biofarmacéutico consistió en control de peso y uniformidad de dosis y ensayo de disolución de acuerdo con las metodologías descritas en la USP XXVI, 20048 para los principios activos seleccionados. La prueba de uniformidad de contenido se realizó mediante: a) el test de control de peso de cápsulas seleccionadas al azar, para lo cual se utilizó una balanza perfectamente calibrada; y b) la valoración de la cantidad de principio activo en cada cápsula. Para la realización de los ensayos de disolución se utilizó un equipo de disolución Erweka modelo DT 600 de 6 posiciones (aparato 2, paleta) y las condiciones del ensayo se ajustaron según el principio activo analizado.
Finalmente, se valoró por una técnica espectrofotométrica la cantidad de principio activo remanente al someter las distintas formas de presentación (tabla 1) a condiciones extremas de almacenamiento, tanto de humedad relativa (HR) 65 % como de temperatura (50 °C). Para tal fin se utilizó una estufa de estabilidad en donde se mantuvo y evaluó periódicamente el contenido en principio activo de las unidades sometidas a estas condiciones, en su envase final.9
Tabla 1: Dosis y tipo de envase utilizado en preparaciones galénicas de Furosemida, Codeína fosfato y Ranitidina clorhidrato
| Dosis/cápsula (mg) | ||||||
| Tipo envase | Ranitidina *HCL | Furosemida | Codeína fosfato | |||
| DU | 5 | 20 | 3 | 10 | 15 | 60 |
| FNU* | 5 | 20 | 3 | 10 | 15 | 60 |
| G | 5 | 20 | 3 | 10 | 15 | 60 |
* Fraccionamiento no unitario (mensual).
G: granel; DU: dosis unitaria; FNU: envase mensual.
Los resultados de uniformidad de peso, contenido y estabilidad fueron evaluados utilizando un paquete estadístico SPSS. Con el objeto de ver si había diferencias significativas en los parámetros estudiados, se aplicó un análisis de varianza (ANOVA) de una vía, considerando el porcentaje de fármaco inalterado como fuente de variación. El nivel de significación usado fue de p < 0,05.
Resultados
Los resultados obtenidos en la prueba de uniformidad de contenido se basan en 2 características de calidad: uniformidad de peso y de contenido por valoración de la cantidad de principio activo por cápsula. Los resultados se muestran en la figura 1 como porcentaje de variación con respecto al peso promedio de las cápsulas y del contenido de los principios activos, obtenido por valoración.
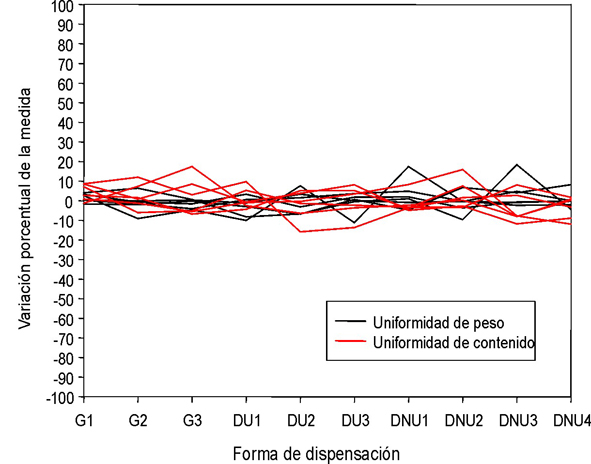
Fig. 1. Variación porcentual de la uniformidad de peso y de contenidos de los principios activos en cápsulas de gelatina dura.
Dada las condiciones de elaboración de la unidad galénica del hospital, para la determinación de la uniformidad de contenido se consideró la cantidad valorada promedio como referencia para el criterio de aceptación o rechazo del ensayo. Cabe destacar, sin embargo, que esta no es una prueba exigible para preparados magistrales terminados, como garantía y control de calidad en el país (propuesta ISP, 2006).10
La variación de peso en ambas dosis de Ranitidina clorhidrato y Codeína fosfato no sobrepasa 10 %, y cumple con lo especificado en la USP. De 20 cápsulas de Furosemida en dosis de 3 y 10 mg analizadas, solo 2 se hallaban entre 15-20 % de variación. La uniformidad de contenido para los 3 fármacos en ambas dosis se halla en torno a 10 % de error relativo, salvo en 3 unidades de un total de 36 ensayadas. En todo caso, el error relativo en el contenido tendió a ser superior para las preparaciones de Codeína fosfato, tanto en altas como en bajas dosis.
Los resultados obtenidos en el test de disolución se basan en la cantidad de principio activo que se encuentra disuelto a los 30 min del ensayo. La especificación dice que 75 % de la dosis debe estar disuelta al cabo de 30 min.
En la tabla 2 se muestra que en promedio, la disolución de los principios activos desde las cápsulas es superior a 90 % para ambas dosis estudiadas.
Tabla 2. Resultados del test de disolución de los principios activos estudiados (porcentaje de fármaco disuelto a los 30 min)
| Nº test | Porcentaje (%) de fármaco disuelto a los 30 min desde cápsulas de gelatina dura | |||||
| Ranitidina*HCL 5mg | Ranitidina*HCL20 mg | Furosemida 3 mg | Furosemida 10 mg | Codeína fosfato 15 mg | Codeína fosfato 60 mg | |
| 1 | 102,2 | 100,6 | 92,0 | 92,6 | 93,1 | 102,9 |
| 2 | 95,5 | 105,7 | 100,7 | 98,0 | 95,2 | 96,8 |
| 3 | 93,3 | 88,0 | 100,7 | 98,5 | 114,0 | 96,4 |
| 4 | 100,0 | 95,4 | 88,0 | 86,6 | 106,2 | 102,1 |
| 5 | 93,3 | 104,0 | 91,3 | 97,4 | 79,3 | 99,3 |
| 6 | 115,6 | 100,6 | 94,9 | 101,5 | 86,9 | 93,9 |
| 7 | 102,2 | 95,4 | 75,6 | 91,4 | 95,2 | 101,2 |
| 8 | 93,3 | 105,7 | 103,3 | 95,4 | 80,7 | 93,9 |
| 9 | 95,5 | 85,1 | 93,8 | 102,8 | 95,2 | 93,9 |
| 10 | 80,0 | 86,3 | 97,5 | 93,0 | 77,2 | 93,6 |
En la figura 2 se puede observar el porcentaje de fármaco activo remanente en función del tiempo en las distintas formas de envasado empleadas para Furosemida a 2 dosis (se omiten los resultados con los otros 2 principios activos, dado que poseen similar tendencia). Con fines de comparación, para evaluar el efecto del sistema de envase sobre la estabilidad de los fármacos en función del tiempo de exposición a alta temperatura y humedad relativa, se realizó un análisis de varianza de una vía con un nivel de significación de p < 0,05.

Fig. 2. Porcentaje de fármaco activo en función del tiempo y de la modalidad de envase (furosemida).
El análisis estadístico arrojó que la estabilidad de cualquiera de los principios activos no depende de la forma de presentación empleada. Sin embargo, cualitativamente, los gráficos muestran una tendencia de mejor conservación de los principios activos cuando estos se envasan en recipiente unitario con papel termosensible.
Discusión
Los resultados de uniformidad de dosis pueden deberse al cambio reciente del método de llenado de cápsulas duras desde una modalidad manual hacia una semiautomática, lo que puede explicar los resultados satisfactorios. El procedimiento de mezclado aún se realiza en forma manual, pero se ha mejorado con la incorporación de un molinillo eléctrico, que ha permitido homogeneizar el tamaño de las partículas; esto podría explicar también los resultados satisfactorios de la uniformidad de la dosis en los 2 extremos estudiados. Estos estudios confirman que el procedimiento, así como el diluyente empleado, no interfieren con el proceso de disolución de los principios activos. Asimismo, se podría extender esta conclusión hacia otras formas galénicas sólidas que se elaboran en la misma unidad con igual procedimiento y excipientes de dilución. Con respecto a la estabilidad, una recomendación que nace de este estudio sería el considerar el envase unitario como forma de acondicionamiento para otras formas galénicas elaboradas en esta unidad, dado sus mejores características de resistencia hacia el efecto de la temperatura y humedad.
Evaluation of the biopharmaceutical characteristics and the stability of solid galenic preparations
Summary
Some biopharmaceutical characteristics such as disolution and uniformity of the content of galenic forms of three active labile principles were evaluated, as well as their stability under conditions of thermal stress and relative humidity in the different containers used. To meet the need of specific pediatric or patient doses that are not available as commercial products, a common practice of the hospital is to make and fraction doses as galenic preparations. The pharmacy of Dr. Gustavo Fricke Hospital, in
Key words: Biopharmaceutical quality, stability, hard capsules, fractioned doses.
Referencias bibliográficas
1. Salgado AC, Rosa ML, Duarte, MA, Almeida, JD. Stability of Spirinolactone in an extemporaneously prepared suspension: the importance of microbiological quality of compounded pediatric formulations. Eur J Hosp Pharm Sci 2005;11(3):68-73.
2. Allen L. Compounding, stability and beyond-use dates. Current & practical compounding information for the pharmacist. Secundum Artem 2003;7(3):1-5.
3. Teraoka R, Otsuka M, Matsuda Y: Effects of temperature and relative humidity on the solid state chemical stability of ranitidine hydrochloride. J Pharm Sci 1993;82(6):601-4.
4. Torres L, Pérez M. Estabilidad. En: Aspectos fundamentales de los sistemas farmacéuticos y operaciones básicas. 1a ed. Madrid: Síntesis, S.A.; 2002;347-62.
5. Abdulrahman M, Fahad J, Khalid A, Mohammad S. Furosemide. Anal Prof Drug Sub 1989;18:153-93.
6. Hohnjec M, Kuftinec J, Malnar M, kreblin M, Kajifež F, Nagl A, et al. Ranitidine hydrochloride. Anal Prof Drug Sub 1983;16:533-61.
7. Farid M, Mahmmound H. Codeine phosphate. Anal Prof Drug Sub 1981;10:93-138.
8. United State Pharmacopeia 26 ed.
9. Chafetz L. Practical testing of solid-state stability of pharmaceuticals. J Pharm Sci 1992;81:107.
10. Propuesta de proyecto ISP. Normas de buenas prácticas de elaboración y control de calidad de fórmulas magistrales y preparados oficinales. Departamento de Control Nacional, Sub-departamento Químico Analítico, Unidad de Magistrales, Instituto de Salud Pública de Chile, Ministerio de Salud, Gobierno de Chile, 2006.
Recibido: 7 de noviembre de 2006 Aprobado: 13 de diciembre de 2006.
Dr. Alexis Aceituno. Laboratorio de Investigación y Desarrollo en Tecnología Farmacéutica, Facultad de Farmacia, Universidad de Valparaíso. Gran Bretaña 1111 Playa Ancha, Valparaíso, CP 5000-1.Teléf.: 56-32-250-8123

