










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introducción
La diarrea refractaria o intratable del neonato constituye uno de los retos que se le presenta a la asistencia pediátrica debido a las variadas etiologías y a las complejidades de su manejo. Se presenta con deposiciones frecuentes, de consistencia blanda y de más de dos semanas de duración, que evolucionan de manera refractaria a los tratamientos habituales.1
Solo el 1 % de los pacientes hospitalizados por diarrea prolongada evolucionan de manera refractaria y se cree que esta condición clínica se relaciona con profundas modificaciones en la función intestinal, muchas veces independientes de la causa inicial y del tratamiento efectuado. Gran parte de los casos permanecen sin diagnóstico y la tasa de mortalidad es elevada.2
Una de las causas de diarrea refractaria es la enteropatía en penacho, conocida como displasia intestinal. Es una condición patológica del intestino extremadamente rara, pero cuando se presenta evoluciona de manera desfavorable en la mayoría de los pacientes.3
El objetivo del presente trabajo es describir el primer reporte en Cuba de enteropatía congénita en penachos, con el fin de llamar la atención de los pediatras y neonatólogos sobre la importancia de estimar este padecimiento en el diagnóstico diferencial de la diarrea refractaria en el lactante y desarrollar un mejor manejo de los casos sospechosos.
Presentación de caso
Paciente nacido vivo, a las 40,2 semanas, con los siguientes antecedentes: tiempo de ruptura de membranas al nacer, líquido amniótico claro, presentación cefálica, parto distócico por cesárea anterior, Apgar 9/9, peso al nacer 3250 gramos, sin antecedentes patológicos personales, serología VDRL no reactiva, grupo sanguíneo O y factor Rh+. No se recogieron los antecedentes prenatales.
Se trasladó de la sala de puerperio al servicio de Neonatología por presentar un vómito, con abdomen distendido y posición en opistótonos. Se realizó perfil de sepsis, con resultado negativo, ultrasonido y radiología de abdomen simple y de pie en los que se observaron edemas interasas. Se inició tratamiento antimicrobiano con cefotaxima, amikacina y metronidazol.
Se decidió realizar tacto rectal con salida de tapón de meconio de 3 cm, no se descartó enfermedad de Hirschprung o megacolon íleomeconial. A las 60 horas de vida presentó aumento de la distensión abdominal y a pesar de no encontrarse obstruido, necesitó estimulación para defecar.
A los cuatro días de nacido se admitió en el Servicio de Cirugía Neonatal para continuar estudios, a pesar de que comenzó a defecar espontáneamente. A los 12 días de edad se trasladó al Hospital Pediátrico Provincial Mártires de Las Tunas, por desarrollar cuadro diarreico con deshidratación severa, necesitando ventilación. Se instauró terapia antimicrobiana con ceftriaxona y vancomicina, manteniéndose bajo ventilación sin controlar cuadro diarreico y no toleró la vía oral. Se realizó hemocultivo en el cual se aisló Staphylococcus aureus, resistente a la vancomicina, con la cual cumplió seis días y se inició tratamiento con linezolid y meropenem. A los 23 días de edad se retiró la ventilación, solo para ser reinstituida poco después debido a la aparición de abundantes secreciones respiratorias espesas y blanquecinas, con elementos de atelectasia. Las deposiciones diarreicas continuaron, con presencia de flemas, fluctuando en intensidad de un día a otro. En los cultivos de las secreciones respiratorias se aisló Escherichia coli. Con este cuadro complejo, a los 38 días de edad el paciente se trasladó a la Unidad de Cuidados Intensivos Pediátricos.
Se discutió el caso en colectivo por neonatólogos, pediatras intensivistas, genetistas clínicos, neurólogos, grupo de fibrosis quística, gastroenterólogos y cirujanos pediatras, coincidiendo todos en la presencia de signos de daño cerebral de causa prenatal, por lo que se ordenaron estudios metabólicos, citogenéticos y de electrólitos en sudor. Además, se administraron probióticos, a fin de recuperar la flora intestinal tan dañada por la terapia antimicrobiana. Debido a las intensas diarreas y al estado séptico severo el paciente desarrolló una desnutrición proteico-energética importante.
En el examen físico se encontraron:
Mucosas húmedas y normocoloreadas
Tejido celular subcutáneo: edema blando de fácil godet en el dorso de miembros inferiores
Piel rosada pálida con úlceras en occipucio y sacras, con herida en región inguinal izquierda debida a venodisección abierta limpia
Respiratorio: polipnea con ruidos transmitidos, murmullo vesicular rudo con subcrepitantes. Tiraje intercostal de dos puntos, frecuencia respiratoria 70 respiraciones por minuto
Cardiovascular: ruidos rítmicos de buen tono e intensidad, con soplo sistólico II/VI aórtico, frecuencia cardiaca de 146 latidos por minuto. Llenado capilar de dos segundos y pulsos presentes en todos los miembros
Abdomen blando, depresible, distendido, con ruidos hidroaéreos presentes y normales, sin visceromegalia. Se reportó además presencia de rectorragia
Sistema nervioso: fontanelas amplias y suturas diastásicas, con respuesta a los estímulos
El paciente se deterioró, presentó hemodinamia insuficiente, con aminas vasoactivas, oxígeno suplementario, estando hipotenso y bradicárdico. Se transfundió en varias ocasiones y se repuso volumen, pero a los 41 días de edad cayó en parada cardiorrespiratoria que no se logró revertir y falleció.
Se realizó la autopsia y el material se fijó en formalina al 10 %. Se procesó con los métodos de rutina y se realizaron coloraciones con técnicas de hematoxilina-eosina. El informe de la autopsia describe:
Hábito externo: cadáver de lactante de 41 días de nacido, masculino constitucionalmente microsplácnico, en estado de conservación, que impresiona desnutrición severa
Cabello ralo, normoimplantado, se observa úlcera por presión incipiente en occipucio
Macizo facial con acentuación de las eminencias óseas, cuello delgado
Tórax con acentuación del relieve de las costillas. Se observaron algunas hemorragias petequiales en hemitórax derecho. Abdomen globuloso, extremidades con múltiples punturas y hematomas recientes. Marcada reducción del panículo adiposo. Cavidades esplácnicas con órganos in situ, libres de líquidos patológicos
Hallazgos anatomopatológicos macroscópicos:
Pulmones rojo-violáceos, pesados y tumefactos (distress), cisuritis interlobar, pleuritis fibrinosa, focos hemorrágicos hacia lóbulo inferior de pulmón izquierdo, traqueobronquitis aguda supurada
Corazón: hemopericardio y discreta dilatación de ventrículo izquierdo
Aparato digestivo: esofagitis aguda, impregnación biliosa de la mucosa gastroduodenal, hepatosplenitis reactiva, hígado de aspecto colestásico, íleon con molde hemorrágico, con mucosa edematosa y congestiva, con dilatación moderada y aspecto atenuado de la mucosa, colon con haustras atenuadas y mucosa congestiva
Riñones: palidez de las pirámides de Malpighi. Riñón izquierdo de mayor tamaño con doble emergencia del sistema excretor. Riñón derecho con hidronefrosis ligera, con área de constricción ureteral a 4 cm de la pelvis y atrofia cortical moderada
Hallazgos histopatológicos:
Pulmones: elementos de edema pulmonar de permeabilidad, dados por membranas hialinas adosadas a la pared septal, con áreas de engrosamiento de los tabiques alveolares con hipercelularidad intersticial
Bronconeumonía multifocal bilateral, con colonias bacterianas basófilas en las luces bronquiolares y presencia de numerosos macrófagos alveolares
Tromboembolismo pulmonar séptico de ramas finas
Hemorragias pulmonares difusas bilaterales
Aparato digestivo: acantosis del epitelio esofágico con congestión aguda.
Yeyuno e íleon con atrofia vellositaria grado IV parcelar con distorsión arquitectural, formación de penachos epiteliales, más prominentes en las puntas vellositarias, como agregados de enterocitos displásicos con redondeamiento de la membrana apical, que ofreció la apariencia de desprendimiento enterocítico. Hiperplasia de las criptas con formación de penachos hacia la luz y dilatación pseudoquística de algunas de ellas. Reducción de la densidad celular del tejido linfoide submucoso y del infiltrado de la lámina propia de la mucosa con importante depleción de la población de células plasmáticas (Figs. 1y 2)
Páncreas: dentro de límites histológicos
Hígado: esteatosis hepática macrovesicular, colestasis canalicular y sinusoidal, necrosis hepatocitaria multifocal con escasos neutrófilos
Bazo: depleción marcada de los manguitos periarteriolares, observándose algunas arteriolas peniciliares desnudas (disreactividad inmunológica)
Riñones: elementos de necrosis tubular aguda
Glándulas suprarrenales: depleción lipídica cortical
Encéfalo: edema cerebral severo
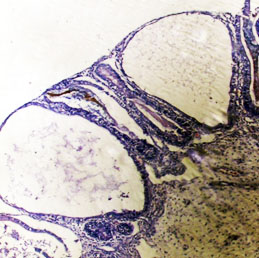
Fig. 1 Micrografía de fragmento de mucosa duodenal teñido con hematoxilina y eosina (H/E), muestra atrofia vellositaria subtotal (enteropatía grado 4) con marcada dilatación pseudoquística de las criptas de Lieberkhün.
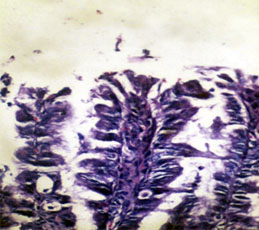
Fig. 2 Micrografía de fragmento de mucosa duodenal teñida con H/E en la que se observan vellosidades con penachos epiteliales prominentes, con aglomeración apical de enterocitos displásicos.
Los hallazgos histopatológicos (con coloración rutinaria de hematoxilina y eosina) de yeyuno e íleon, fueron consistentes con el diagnóstico de enteropatía congénita en penachos (Fig. 1).
Se decidió realizar análisis inmunofenotípico, se enviaron las muestras de tejido fijado en formol y embebido en parafina al Centro Nacional de Referencia en Anatomía Patológica en el Hospital Clínico Quirúrgico Hermanos Ameijeiras. Se determinó CD10, CEA, CD4, CD8 y CD3.
Los especímenes de yeyuno sometidos al estudio inmunohistoquímico exhibieron inmunorreactividad para CEA y CD10, con patrón de tinción lineal ininterrumpida de la membrana apical de los enterocitos, corroborando el diagnóstico en cuestión. Los marcadores CD3, CD4 y CD8 permitieron evaluar las características de la población linfoide, depletada y de predominio T (Fig. 3).
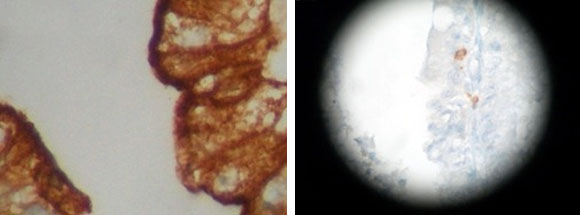
A. Inmunotinción con CEA (antígeno carcinoembrionario) que exhibe patrón de tinción lineal ininterrumpida de la membrana apical de los enterocitos | B. Depleción linfocitaria, en la micrografía solo se advierten 2 linfocitos TCD4+ con migración transepitelial.
Fig. 3 Micrografía de fragmento de mucosa duodenal.
Discusión
La enteropatía en penachos fue descrita por Reifen y otros en el año 1994.1,3 En Europa se estima que la incidencia es de 1 en 50 000 a 100 000 nacidos vivos. La enfermedad se identifica como trastorno mendeliano con patrón de herencia autosómico recesivo. Aunque no exclusivamente, se reporta en poblaciones con tendencia a la endogamia y a matrimonios consanguíneos.1,4
Desde el punto de vista clínico, se presenta en el periodo neonatal, con un cuadro diarreico lo suficientemente severo como para requerir soporte nutricional, especialmente de tipo parenteral. La diarrea suele persistir en forma leve, a pesar del reposo intestinal. Cuando es masiva puede llevar a la deshidratación y a disturbios hidroelectrolíticos de forma rápida, con la consecuente descompensación hemodinámica.1,5) Características descritas en el presente caso.
Este padecimiento presenta heterogeneidad clínica e histológica y puede asociarse, entre otras alteraciones, con malformaciones como rasgos dismórficos faciales, micrognatia y ano imperforado.6Aunque no está totalmente esclarecida su base molecular, existen evidencias de su relación con anomalías del desarrollo de la membrana basa, como depósitos débiles e irregulares de laminina en la interfase entre la lámina propia y el epitelio y de heparán sulfato como depósitos laminares.1,7,8,9
Además, se describen cambios ultraestructurales en los desmosomas, como disrupción y contactos mediados por integrinas, sugiriendo una analogía fisiopatológica con la epidermólisis bullosa cutánea. También se observa una distribución anormal de α1β2 integrinas, sugiriendo que los cambios podrían resultar de una interacción anormal entre las células o entre la célula y la matriz.1) Muchos de los casos permanecen sin diagnóstico y la tasa de mortalidad es elevada.1,7
El diagnóstico es histológico, por lo que se necesita una biopsia duodenal. En esta se evalúa de forma protocolizada la relación vellosidad/cripta, la presencia de hiperplasia de las criptas, el componente inflamatorio de la lámina propia e intraepitelial, considerando el tipo de células presentes como linfocitos, células plasmáticas y eosinófilos.2,5,7
Los hallazgos característicos de la afección consisten en la presencia de proyecciones irregulares en forma de penachos epiteliales, con discohesividad enterocítica, dilatación pseudoquística de las criptas, con grados variables de atrofia vellositaria (atrofia parcial leve a subtotal) y un patrón de tinción lineal de la membrana apical de los enterocitos con coloraciones histoquímicas como el ácido Peryódico de Schiff e inmunohistoquímicas como CEA y CD10.1,2,10
El diagnóstico diferencial se realiza con el resto de las enfermedades que se presentan con diarrea refractaria neonatal y atrofia intestinal, especialmente, la de inclusión de microvellosidades y la enteropatía autoinmune. En la primera, la tinción membranosa es discontinua y, mediante el análisis ultraestructural, se revela una marcada vesiculación subapical. En la enteropatía autoinmune se verifica una severa inflamación destructiva con marcada atrofia vellosa y críptica, numerosos focos de apoptosis con satelitosis linfocítica hacia la base de las criptas1,7
Puede requerirse la repetición de las biopsias, demorando el diagnóstico debido a que la presencia de los penachos parece variar con el tiempo y distribuirse en forma de parche.2) Para el manejo de la afección se requiere del empleo de medidas especiales de soporte hidroelectrolítico y nutricional para el sostén homeostático del neonato. La alimentación enteral continúa con fórmulas hidrolizadas de proteínas o aminoácidos que, por periodos prolongados, empeoran la diarrea, por lo que la mayoría de las veces se utiliza nutrición parenteral. En algunos casos se precisa de trasplante intestinal, indicación que se debate al considerar los riesgos de trombosis y sepsis de la nutrición parenteral prolongada.11
Los primeros estudios moleculares realizados en enteropatía en penachos relacionaba al gen EpCAM (codifica para molécula de adhesión de células epiteliales) como causa de la enfermedad.12,13,14 Recientemente, se identificaron 42 mutaciones diferentes de EpCAM. Estas mutaciones conducen a deleciones en los cromosomas, empalmes defectuosos, cambios de marco de lectura, truncamientos y mutaciones silentes.15,16 Está descrito que pacientes con mutaciones de cambio de marco de lectura o truncamiento en el gen EpCAM (c.499dupC) producen genotipos con efectos más graves de la enfermedad.17 Es válido destacar que, cuando el gen EpCAM se encuentra mutado o truncado, la proteína resultante disminuye en abundancia y se encuentra mal localizada en la célula.18
Más recientemente, se constató que mutaciones del inhibidor de la serina peptidasa Kunitz tipo 2 (SPINT2) también están involucradas con la ocurrencia de la enfermedad.6,19) Varios modelos con mutaciones SPINT2 mostraron evidencia de un síndrome similar a la enteropatía congénita y alteración de la homeostasis intestinal.2,19 Dichos mutantes provocan la disminución/pérdida de EpCAM a través de la actividad matriptasa, que finalmente conduce a la aparición de la enfermedad. No obstante, la mayoría de los pacientes con enteropatía en penachos poseen mutaciones en el gen EpCAM. Una revisión reciente sugiere que el 74 % de los pacientes con la enfermedad portan mutaciones en EpCAM, mientras que el 26 % tienen mutaciones en SPINT2.2) Estos hallazgos permitirán aumentar el conocimiento sobre las bases moleculares de la enteropatía en penachos.19
El caso que se presenta tuvo características clínicas e histopatológicas congruentes con lo que se describe en la literatura internacional, con diagnóstico tardío y evolución letal. Además, es el primer reporte en Cuba a partir de hallazgos histopatológicos, con lo cual se llama la atención hacia la presencia del genotipo mórbido del padecimiento. Desafortunadamente, el diagnóstico molecular de la enfermedad no estuvo a nuestro alcance.
En resumen, la enteropatía en penachos supone un reto diagnóstico al no exhibir un cortejo clínico patognomónico. La concomitancia de diarrea crónica con los trastornos malformativos debe hacer saltar las alarmas y orientar el pensamiento clínico y la metodología diagnóstica hacia posibles trastornos genéticos, en los que se requiere biopsia intestinal oportuna con mapeo amplio, para el diagnóstico temprano y conducta más favorable.

