










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Oftalmología
On-line version ISSN 1561-3070
Rev Cubana Oftalmol vol.20 no.2 Ciudad de la Habana July-Dec. 2007
Centro de Referencia Nacional de Retinosis Pigmentaria
Hospital Docente “Dr. Salvador Allende”
Características clínicas y evolución de la retinosis pigmentaria en adolescentes
Raisa Hernández Baguer,1 Mirta Copello Noblet,2 Ana María Cabezas García,3 Daysi Domínguez Rodríguez3 y Bárbara Cid Vázquez4
Resumen
Objetivo: Identificar las características clínicas y la evolución de la retinosis pigmentaria en la adolescencia.
Método: Estudio descriptivo y prospectivo en adolescentes entre 10 y 14 años de edad (grupo I) y entre 15 y 19 años, (grupo II), del sexo masculino y del femenino, atendidos en Centro de Referencia Nacional de Retinosis Pigmentaria- Pediátrico entre 2002 y 2006. Se observó herencia, síntoma inicial y edad, tiempo transcurrido y enfermedades generales concomitantes y los signos del FO por biomicroscopia y oftalmoscopia, perimetría y ERG al inicio, anual y al final. Se clasificó el estadio en 4 etapas atendiendo a parámetros como agudeza visual con corrección. y CV.
Resultados: Entre los 42 adolescentes predominaron los varones y el color de la piel blanca. El DVP estuvo presente en 12 ojos (14,2 %), las opacidades del cristalino en 61 ojos (71,4 %), principalmente en el grupo 2. Al final del estudio, según la agudeza visual y CV, 32 adolescentes (76,2 %) tuvieron con evolución no progresiva, en 5 (11,9 %) fue medianamente progresiva y en 5 (11,9 %) fue muy progresiva pues hubo marcado empeoramiento de la función visual.
Conclusiones: Las características de la retinosis pigmentaria que se identificaron en los adolescentes se incluyen entre los 3 primeros estadios. La evolución durante 5 años no fue progresiva en la mayoría de los adolescentes de ambos grupos, lo cual induce a considerar que los cambios biológicos de esta etapa de la vida aparentemente no influyen en el curso de la retinosis pigmentaria.
Palabras clave: Retinosis pigmentaria, adolescencia, ceguera nocturna.
La edad de la adolescencia es un período de la vida del ser humano caracterizado por rápidos y diversos cambios que se suceden de forma muy dinámica, cambios físicos y emocionales fundamentales, que crean las características de la vida adulta ulterior.1,2
Una edad tan compleja y trascendental requiere también una atención consecuente. De un modo convencional se considera que la adolescencia es el período que cubre la segunda década de la vida (desde 10 hasta 20 años). No existe otro momento después del nacimiento en que los hechos biológicos se produzcan tan rápido.
En esta segunda década de la vida aparecen enfermedades oculares de tipo distrófico genéticamente determinadas, cuya naturaleza familiar ha sido conocida por más de 100 años.
Entre estas enfermedades oculares distróficas de evolución crónica y de origen genético se encuentra la retinosis pigmentaria (RP), terminología que agrupa una serie de enfermedades que de forma primaria y progresiva afectan los fotorreceptores (conos y bastones) y después otras capas de la retina ocasionando ceguera nocturna, disminución de la visión periférica y posteriormente también de la visión central dando lugar a una discapacidad visual con características muy particulares. La retinosis pigmentaria constituye la primera causa de ceguera de tipo distrófico y hereditaria.3
Otra característica clínica a destacar es la posible concomitancia de la RP con enfermedades generales endocrinas, trastornos auditivos, neurológicos, etc., que conforman verdaderos síndromes. Se trata entonces de una RP asociada.3
En su curso natural la RP puede debutar a cualquier edad y llegar hasta la ceguera total en un tiempo variable -esto dependiente un tanto de la edad de comienzo y de la severidad del tipo de RP-; sin embargo, la literatura mundial muy poco se ha referido a las características de la enfermedad durante la etapa de la adolescencia.4
La prevalencia mundial de esta enfermedad se considera de 1 x 5 000 habitantes, y se cree que 1 de cada 80 personas presenta el gen causante de la enfermedad.5 En nuestro país por las particularidades del Sistema Nacional de Salud y por la pesquisa que se despliega en la población cubana por la red nacional del Programa de Atención a Pacientes con Retinosis Pigmentaria y familiares conocemos que la prevalencia es de 4,1 x 10,000.6
Debido a las limitaciones que la RP le ocasiona al individuo que la padece, ella repercute indiscutiblemente en su estilo y calidad de vida.
Por lo anteriormente expresado la RP se ha insertado como un problema prioritario de salud en el marco de nuestro pesquisaje activa de enfermedades visuales en mayores de 5 años y en el programa de salud para discapacitados que no tiene igual en el mundo. El objetivo de este estudio nos permitió ganar en información y comprensión acerca de este problema de salud en cuanto su comportamiento en la etapa de la adolescencia, es decir, identificar sus características clínicas y su evolución.
Métodos
Se realizó un estudio descriptivo, longitudinal y prospectivo en adolescentes entre 10 y 19 años de edad (según criterio de la OMS) atendidos en el Centro de Referencia Nacional y el Pediátrico de Retinosis Pigmentaria (Cuba) desde el año 2002 hasta el año 2006.
Se recogió en encuestas individuales los datos que permitieron determinar las características clínicas de la enfermedad en cada adolescente, el resultado de los exámenes oftalmológicos al inicio, anualmente y al final del estudio durante 5 años para después de procesados estos, ofrecer los resultados correspondientes según los objetivos de la investigación. Para determinar los antecedentes clínicos y las características de la RP se tuvieron en cuenta: tipo de herencia, síntoma inicial y edad en que se manifestó, tiempo de evolución de la enfermedad ocular, enfermedades generales y oculares concomitantes y los signos en el fondo de ojo que definen el tipo de RP de que se trata. Se confirmó el diagnóstico por electrorretinograma con equipo computadorizado EREV 2000 Plus de la LACE y se realizó perimetría cinética tipo Goldmann con estímulo blanco de 64 mm² y filtro de 1.00 (Suma V/4)
Se clasificó el estadio clínico de la enfermedad en 4 etapas atendiendo a parámetros como agudeza visual con corrección y área de campo visual residual, teniendo como elementos colaterales a considerar el estado de los medios transparentes y la descripción detallada de la estructuras del fondo de ojo por biomicroscopia anterior y posterior y oftalmoscopia directa e indirecta respectivamente.
La clasificación responde a la metodología de trabajo de la Escuela Cubana de RP fundada por el Dr.CM, Profesor Orfilio Peláez Molina † (1923-2001) en 1989 utilizada para evaluar estos parámetros principales. Los estadios clínicos son:
- I con AV ≥ 0,5 y CV ≥ 15,°
- II con AV ≥ 0,3 y CV entre 11° - 15°,
- III con AV ≥ 0,05 y CV entre 5° - 10° y
- IV con AV ≤ 0,05 y CV – de 5° considerando el ojo mejor.7
Para identificar la forma de evolución de la RP se tomó en cuenta el estadio clínico de la enfermedad en cada adolescente, al inicio y al final del estudio y se tuvo en cuenta cada tipo de RP (típica, atípica o asociada) y de herencia (autosómica dominante, autosómica recesiva, o recesiva ligada al cromosoma X).
Se establecieron 3 formas de evolución:
- No progresiva: No hubo cambio de estadio clínico.
- Medianamente progresiva: Hubo cambio al estadio clínico siguiente de la clasificación.
- Muy progresiva: Hubo cambios a estadios más avanzados de la clasificación.
Esta calificación nos permitió conocer el comportamiento de la RP en cada uno de los jóvenes después de transcurridos 5 años del estudio comprendidos dentro de la etapa de la adolescencia y de determinar la posible influencia de los cambios biológicos y psíquicos de esta etapa sobre el curso de la enfermedad ocular.
Se obtuvo la distribución de acuerdo con cada variable, que se definieron utilizando como medida de resumen el porcentaje, y se expresaron los resultados en tablas y figuras.
Resultados y discusión
Las características de la muestra estudiada en cuanto a edad y sexo se muestran en la figura 1. Los adolescentes de nuestro estudio (42) se distribuyeron en dos grupos (I y II), según la edad en años, al inicio del estudio, en el año 2002. (Predominaron los varones.)
- Grupo I con 10 años de edad (correspondían a la adolescencia temprana).
- Grupo II con 15 años de edad (correspondían a la adolescencia media hasta tardía),8 pues se requería que aún fueran adolescentes al final del estudio, una vez transcurridos 5 años.
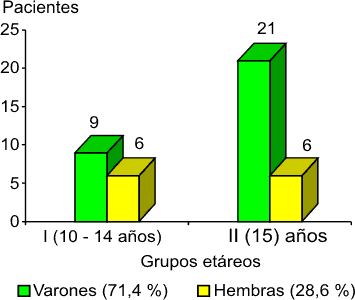
Fig. 1. Retinosis pigmentaria y adolescencia. Distribución por edad y sexo.
En relación con el color de la piel se reporta que la RP afecta de forma mayoritaria a los individuos de la raza blanca y a los varones.6,9 De igual forma se comportó en nuestra muestra para un 61,9 % del tipo caucásico.
Atendiendo a los antecedentes clínicos de la RP, la edad en que apareció el primer síntoma, es decir la ceguera nocturna, señalado como debut de la enfermedad, fue precoz antes de 10 años de edad (30,9 %) y juvenil de 10 años en adelante (69,1 %).7 Al precisar el debut resultó que el tiempo de evolución de la enfermedad estuvo entre 5 y 10 años al inicio del estudio para algunos pacientes ya diagnosticados como resultado de la pesquisa familiar realizada durante años por el programa nacional cubano.
Nuestros adolescentes habían presentado diferentes enfermedades generales antes del inicio del estudio, lo cual nos fue informado al interrogar los APP; otras afecciones ocurrieron durante la investigación. La tabla 1 refleja solamente aquellas enfermedades que nos interesaron porque a veces forman parte de síndromes conocidos. El determinar si la RP es parte de un síndrome será consecuencia del estudio y de la observación del paciente durante años. Otros padecimientos como afecciones ortopédicas, estomatológicas, psicológicas, infectocontagiosas, dermatopatías, problemas ginecológicos, etc., propios de esta etapa de la vida10 fueron debidamente recogidas en encuestas individuales y atendidos como está orientado en el Programa Nacional de Atención Integral a la Salud de Adolescentes (2000) del Ministerio de Salud Pública de Cuba.
La asociación de RP e hipoacusia neurosensorial ocasionando la doble discapacidad de sordoceguera apuntó hacia la presencia de síndrome de Usher11 en 3 pacientes y 1 con hipoacusia por ototoxicidad medicamentosa (tabla 1).
El hábito de fumar estaba presente en 7 adolescentes varones (16,6 %) que formaban parte del grupo II.
Tabla 1. Retinosis pigmentaria. y adolescencia. Enfermedades generales asociadas
| Enfermedades asociadas | Número | % |
| Hipoacusia | 4 | 9,5 |
| Asma | 9 | 21,4 |
| Epilepsia | 2 | 4,7 |
| Hipertensión arterial | 2 | 4,7 |
| Sin enfermedad | 30 | 71,4 |
Nota: Puede aparecer más de una enfermedad por adolescentes. % calculado del total de adolescentes.
Todos estos adolescentes (42) se atienden en los Centros de Referencia Pediátrico y Nacional, una vez confirmado el diagnóstico de RP han recibido el tratamiento cubano para esta enfermedad, esquema terapéutico que comprende ozonoterapia, electroestimulación, vitaminoterapia y cirugía revitalizadora. Este proceder corresponde a la metodología de trabajo de los centros de la Red Nacional de Retinosis Pigmentaria en Cuba.12 (Consideramos oportuno hacer esta aclaración aunque el evaluar el resultado del tratamiento médico aplicado no es el objetivo de nuestra investigación.)
La presencia de pigmentos retinianos, la alteración del epitelio pigmentario dando un aspecto no homogéneo al fondo de ojo como granulación fina y el estrechamiento del calibre vascular son los signos más precoces del RP en el fondo de ojo de nuestros adolescentes con menos tiempo de evolución de la enfermedad.13-15
De forma más tardía aparecieron las variaciones del color del disco óptico y del aspecto de la mácula entre 7 u 8 años de evolución de la enfermedad. Todos estos cambios pueden verse de forma bilateral y bastante simétrica en el fondo de ojo.
Se hallaron otras enfermedades oculares acompañando la RP como las ametropías, el astigmatismo miópico compuesto, el estrabismo latente y nistagmo que siempre tienen alta incidencia generalmente en la etapa preescolar y escolar, por eso están presentes en este grupo poblacional de nuestro estudio.16,17
En el examen de los medios trasparentes la córnea no se presentaron alteraciones, en cambio, las alteraciones vítreas se hallaron más tempranamente que las de cristalino. Se observó celularidad, desorganización fibrilar y flóculos en ese orden de aparición. El desprendimiento de vítreo posterior estuvo presente en 12 ojos (14,2 %), los cambios en la transparencia del cristalino y la catarata pequeña en extensión y de localización polar posterior16,17 estuvieron presentes en 61 ojos al final del estudio principalmente en adolescentes del grupo 2. Estas alteraciones del vítreo y del cristalino no se observan en jóvenes sanos,3,13 por lo cual se confirma que la temprana aparición de ellas en nuestros adolescentes está relacionada con diversos factores en el transcurso de la RP3 (tabla 2) (p < 0,001).
Sería prácticamente imposible observar este resultado si en la RP no se afectaran verdaderamente los medios transparentes. Estudios epidemiológicos (Harding, 1991) señalan el aumento en la incidencia de las opacidades del cristalino en las distintas distrofias vitreorretinianas.
Tabla 2. Retinosis pigmentaria. y adolescentes. Estudios de los medios transparentes
| Estructura | Con alteraciones | Sin alteraciones | ||
| Número de ojos | % | Número de ojos | % | |
| Cristalino | 61 | 72,6 | 23 | 27,4 |
| Vítreo | 28 | 33,4 | 56 | 66,6 |
Nota: Puede aparecer más de una alteración de los medios por ojo y por adolescente.A partir del total de adolescentes fue calculado.
P<< 0,001
La forma clínica típica de la RP (36 pacientes, 85,6 %) fue la predominante en el fondo de ojo -coincide con lo que describe la literatura mundial.3, 16,17
Se evaluaron los cambios de los estadios clínicos y el tipo de RP y de herencia en cada uno de los adolescentes que tuvieron ese comportamiento en el estudio. Esto nos permitió conocer las características de la evolución en cada persona, tipo de RP y de herencia. En la típica la evolución no fue progresiva en 29 pacientes (80,5 %).
Las retinosis pigmentarias clasificadas atípicas fueron apigmentos 2 pacientes y central o inversa en 1 paciente. Solo en 1 de ellos en la de tipo inversa la evolución fue progresiva (66,6 %) por cambios importantes en la agudeza visual central como se reporta en las RP con toma macular temprana .En este caso la herencia no pudo ser definida.
La retinosis pigmentaria asociada (USHER tipos I y II) fue hallada solamente en 3 casos, como también se observa frecuentemente a nivel mundial.11 La evolución de la RP asociada al final del estudio fue progresiva solo en 1 caso.
Finalmente quisimos conocer si existía alguna relación entre la forma de evolución de la RP y el tipo de herencia y observamos que los adolescentes con herencia autosómica dominante mostraron durante el tiempo del estudio, estabilidad de la enfermedad ya que no se encontraron cambios en el estadio clínico inicialmente determinado (fig. 2).
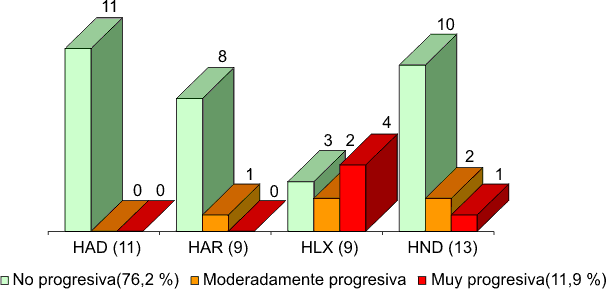
Nota: Porcentaje calculado sobre la base del total de adolescentes (42).
Fig. 2. Retinosis pigmentaria y adolescencia.Características de lña evolución según tipo de herencia.
Algunos autores describen la RP de herencia autosómica dominante como forma benigna de la enfermedad pues en edades avanzadas el enfermo conserva aún buena agudeza visual central y campo visual aceptable para su orientación y desplazamiento.18 En nuestro estudio el pronóstico visual para este tipo de herencia es más favorable.
En la herencia recesiva ligada al cromosoma X, 6 de 9 adolescentes manifestaron progresión de la enfermedad ocasionando cambios en el estadio clínico inicial.
Este tipo de RP se describe como la forma más severa de la enfermedad degenerativa retiniana que conlleva a marcada disminución de la agudeza visual en edades tempranas.19 Nuestros adolescentes con este tipo de herencia se encuentran la mayoría en los estadios clínicos II y III de la RP.
Hubo progresión de la RP también en 3 de 13 adolescentes que no tenían precisado el tipo de herencia, podría tratarse de los patrones autosómico recesivo o recesivo ligados al cromosoma X,18 lo cual no pudo determinarse mediante el estudio del árbol genealógico.
Al final del estudio en relación con la función visual según agudeza y campo visual como criterios mayores, 32 adolescentes (76,2 %) se mantuvieron estables o con evolución no progresiva, en 5 (11,9 %) la evolución fue medianamente progresiva y en 5 (11,9 %), hubo marcado empeoramiento de esta función y por tanto la evolución de la RP fue muy progresiva (fig. 2). En un estudio realizado por Berson durante 3 años de observación del curso natural de la enfermedad en 24 pacientes de 6 a 17 años encontró la pérdida del área del campo visual 4.6 % por año en todos los casos estudiados marcando una progresión de la enfermedad mucho mayor que la observada en nuestros casos.4
El resultado de las tonometrías arrojó una presión intraocular (PIO) dentro de límites normales en aquellos jóvenes en que su cooperación permitió la realización de este examen. Recordemos la posible asociación de RP y glaucoma en el 3 % de los afectados por esta degeneración retiniana.20
Conclusiones
- Las características de la retinosis pigmentaria, observadas en los adolescentes de ambos grupos, se identificaron entre las descritas para los 3 primeros estadios de nuestra clasificación evolutiva de esta enfermedad ocular.
- Esto fue apreciado en la mayoría de los adolescentes tanto para las formas clínicas típica, atípica o asociada, por lo cual consideramos que esto se relaciona con el tiempo de evolución de la enfermedad.
- La evolución de la RP durante el tiempo del estudio (5 años) no ha sido progresiva en la mayoría de los adolescentes del grupo I ni en los del grupo II; lo cual nos permite considerar que los importantes cambios biológicos que forman parte de la adolescencia al parecer no influyen en el curso de la retinosis pigmentaria en esta etapa de la vida.
- La relación entre formas de evolución y tipo de herencia fue significativa en los adolescentes hombres con herencia recesiva ligada al cromosoma X donde hubo empeoramiento de la agudeza y campo visuales en todos ellos sin existir otra causa ocular o causa extraocular que explique este acontecimiento. Esto nos sugiere que existe una evolución de la enfermedad muy particular para este tipo de herencia.
Recomendaciones
- Efectuar la captación temprana de los adolescentes con factores de riesgo dentro de familias afectadas con retinosis pigmentaria para un diagnóstico precoz de esta enfermedad ocular.
- Estrechar los vínculos entre comunidad y familia para la atención integral de los adolescentes que padecen esta enfermedad ocular discapacitante.
Summary
Clinical characteristics and evolution of retinitis pigmentosa in adolescents
Objectives: Identify the clinical characteristics and evolution of retinitis pigmentosa in adolescents.
Method: A prospective descriptive study of adolescents aged 10 to 14 years (group I) and 15-19 years (group II) of both sexes and treated at the National Reference Center of Pediatric Retinitis Pigmentosa from 2002 to 2006. Inheritance, initial symptoms, age, time elapsed and general concomitant diseases, FO signs by biomicroscopy and ophthalmoscopy, perimetry and ERG at the beginning, annually and at the end were all examined. Four stagings were set up according to parameters such as visual acuity with correction and CV
Results: Males and Caucasians predominated in the 42 studied teenagers. DVP was present in 12 eyes (14.2), crystalline opacities were found in 61 eyes (71.4%) mainly in the group II. At the end of the study, according to the visual acuity and CV, 32 adolescents (76,2%) did not have progressive evolution of the disease, 5 (11,9%) had moderately progressive and 5 (11,9%) showed very progressive evolution since there was marked worsening of the visual function.
Conclusions: The identified characteristics of retinitis pigmentosa in adolescents were within the three first stagings. Evolution was not progressive for 5 years in most of adolescents from both groups, which led us to consider that the biological changes in this period of life seem to have no influence in the course of retinitis pigmentosa.
Key words: Retinosis pigmentosa, adolescence, night blindness
Referencias bibliográficas
1. Canessa, Nykiel. Manual para la educación en salud del adolescente. OPS/FNUAP; 2002. No.12.
2. Consuegra R. Problemas médicos de los adolescentes. Ciudad Habana: Científico Técnica; 1999. p 15-38.
3. Gutiérrez Torres SM. Retinosis pigmentaria. Clasificación y tratamiento. 2004 [citado 12 julio 2006]. Disponible en: http://retinois.org / olo / libro / l. htm
4. Berson E, Sandberg MA, Rosner B, Buch DB, Hanson A. Natural course or RP over a three year interval. Am J. Ophthalmol. 1999:240-51.
5. Najera C. Epidemiology of retinitis pigmentosa in the Valencia Community (Spain). Genetic Epidemiology. 1999;37:46.
6. Haim, M. Prevalence of Retinitis Pigmentosa and all disorders in Denmark III Hereditary Pattern. Acta Ophthal. 1998;70:178-86.
7. Bunke CH; Berson BL. Prevalence of Retinitis Pigmentosa in Manila. Am. Ophthal. 1999;11:352-63.
8. MINSAP .Cuba. Programa Nacional de Retinosis Pigmentaria. Reporte Nacional; 2004.
9. Herrera Mora M. Clasificación del profesor Orfilio Peláez Molina. Escuela Cubana de Retinosis Pigmentaria. En: Retinosis Pigmentaria. Experiencia Cubana. Ciudad de La Habana: Científico Técnica; 1997. p.77-81.
10. Peláez Mendoza J. Adolescencia y sexualidad. Ciudad de La Habana: Científico-Técnica; 1996.
11. Pirech. Epidemiology and prevalence of hereditary retinal dystrophies in France. Jour France Ophthal.1997;141(5):153-64.
12. Roselot J. Adolescencia. Problemática de salud del adolescente y joven en Latinoamérica y el Caribe. Pediatría. 2da ed. Menchelo: Intermédica; 1999.
13. Kimberling W. Smith RJH. Gene Mapping of the Usher Syndromes Otolaryngologic Clinic of North America. 2003;25(5):
14. Peláez O. Tratamiento de la retinosisi pigmentaria. cap IX .En: Retinosis pigmentaria. Experiencia cubana .Ciudad de La Habana: Científico Técnica; 1997. p 173.
15. Weleber R.J. Retinitis Pigmentosa and allied disorders. In Ryan S. J. editors. Retina Vol I St. Louis: The CV Mosby Company; 2002. p. 299-315.
16. Gil-Gibernau J. El fondo de ojo en el niño. Ciudad de La Habana: Científico Técnica; 1999.
17. Fondoscopia en la Retinosis Pigmentaria. Mundo Científico Médico. 1998(11):
18. Kanski J.J . Oftalmología clínica. España: Doyma.; 2002.
19. Vaughan O. Oftalmología general. México: El manual moderno; 2002.
20. Kaplan J, Borneau Q. Clinical and Genet Heterogeneity in Retinitis Pigmentosa. Hum. Genet .1998;85:635-42.
21. Miano MG. Mutation analysis of the RPGR gene in south European with X Linked Retinitis Pigmentosa. Biophysics Italy. Euro- JH. Genetics. 2000;7(6):687.
22. Peng T. Wul. Zhou W. Retinitis Pigmentosa Associated with glaucoma. Clinical analysis Eye Sci. 1998;6(1):217-9.
Recibido: 21de abril de 2007. Aprobado: 20 de julio de 2007.
Dra. Raisa Hernández Baguer. Centro de Referencia Nacional de Retinosis Pigmentaria. Hospital Docente “Dr. Salvador Allende”. Calzada del Cerro # 551.Ciudad de La Habana. Cuba. E-mail: raisa.baguer@ infomed.sld.cu
1Especialista de II Grado en Oftalmología. Instructor.
2Especialista de II Grado en Oftalmología. Asistente.
3Especialista de I Grado en Oftalmología. Hospital Pediátrico del Cerro. Calzada del Cerro No. 2002, Ciudad de La Habana, Cuba.
4Licenciada en Tecnología de la Salud.

