










 Custom services
Custom services Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La retinopatía de la prematuridad, conocida por sus siglas en inglés retinopatía de la prematuridad es una enfermedad dinámica vasoproliferativa de la retina inmadura postnatal, incompletamente vascularizada, que afecta a los bebés prematuros.1
Se reconoce por cuatro estadios y un cuadro de mayor gravedad conocido por retinopatía de la prematuridad agresiva posterior. En esta se describen fundamentalmente áreas avasculares de la retina delimitadas de las vasculares, neovascularización, hemorragias, tortuosidad y dilatación de los vasos retinales. En fases más avanzadas puede haber bandas fibrovasculares, ectopia macular y desprendimiento de retina. Considerando el antecedente de prematuridad y los signos típicos de la entidad no resulta difícil para el profesional entrenado diagnosticar pacientes con retinopatía de la prematuridad; sin embargo, pueden aparecer diferentes hallazgos clínicos oculares que semejan o coexisten con dicha entidad, lo que supone un reto diagnóstico para el observador.
En la fase aguda de la enfermedad el diagnóstico diferencial de la retinopatía de la prematuridad implica enfermedades de la retina con un trasfondo genético común en relación con mutaciones de proteínas relacionadas con la llamada vía de señalización de Wnt, la cual desempeña un papel clave en el desarrollo embrionario ocular, específicamente en la angiogénesis retiniana.2,3) La vía de señalización Wnt está altamente conservada y regulada entre muchas especies. Las mutaciones que afectan algunos de los genes de esta vía (NDP, FZD4, TSPAN12 y LRP5) pueden dar lugar a varias vitreorretinopatías pediátricas, como la enfermedad de Norrie, la vitreorretinopatía exudativa familiar y el síndrome de pseudoglioma y osteoporosis. También se han informado mutaciones de la vía
Wnt en la enfermedad de Coats y la vasculatura fetal persistente.4
Como retinopatía de la prematuridad atípica se ha reconocido en la literatura la retinopatía de la prematuridad agresiva posterior, debido a su presentación distinta y la evolución tórpida al demorar el tratamiento, terminando en ocasiones en desprendimiento de retina. Además de la retinopatía de la prematuridad clásica y la retinopatía de la prematuridad agresiva posterior, también se describen pacientes con una forma rara, atípica y grave de retinopatía de la prematuridad en la que cualquier tratamiento establecido no tiene éxito. Otra forma de atipicidad es el desarrollo de proliferación fibrovascular severa en el polo posterior, especialmente en el disco óptico.5
Al realizarle pruebas genéticas a nueve pacientes con retinopatía de la prematuridad atípica que se diagnosticaron durante tres años en un hospital en China se determinó que todos presentaron mutaciones en los genes de la vía de señalización de Wnt.6
Presentación del caso
Se presenta un paciente nacido el 17 de diciembre de 2020 a las 31,1 semanas de gestación con 1 100 g de peso, sexo masculino y color de la piel mestiza. Durante la gestación la madre presentó diabetes gestacional e hipertensión arterial, esto último condujo a la interrupción del embarazo por cesárea. El puntaje del Apgar al nacer fue 8/8, requiriendo oxigenación durante 12 días, de los cuales cinco fueron en modalidad ventilación obligatoria sincronizada intermitente y el resto en ventilación nasal no invasiva. Presentó trastornos de la coagulación, síndrome de distrés respiratorio y requirió antibioticoterapia por sepsis respiratoria.
Recibió su primera evaluación oftalmológica a las 35,2 semanas, como parte de la evaluación establecida en el Programa Nacional de Retinopatía del Prematuro, donde se constata en el ojo derecho una línea demarcatoria en zona 2-3, sin signos de enfermedad plus, ni hemorragias (estadío 1, zona 2-3). En el ojo izquierdo por el contrario, los vasos retinales no sobrepasan la zona 1, y en estadío 3 dado por la existencia de neovascularización, vaso demarcatorio circunferencial y hemorragias en casi todos los sectores (Fig. 1). No se precisa enfermedad plus, pero sospechamos la presencia de una retinopatía de la prematuridad agresiva posterior.
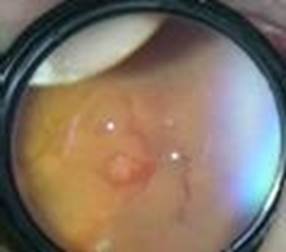
Fig. 1 Imagen de Fondo de ojo en región temporal del ojo izquierdo donde se observa la detección del crecimiento de los vasos en zona 1, neovascularización y vaso demarcatorio.
Se decidió reevaluar en siete días, donde se constata empeoramiento del cuadro: mayor congestión en el extremo distal de los vasos, sin evidencia de crecimiento vascular (Fig. 2). Como elemento añadido es la presencia de exudados intraretinianos y subretinianos en todos los sectores.
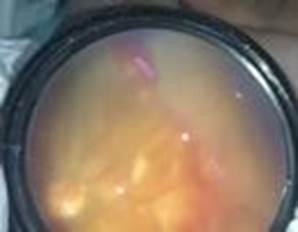
Fig. 2 Imagen de fondo de ojo en temporal ojo izquierdo con mayor congestión de los vasos, sin sobrepasar la zona 1 y presencia de exudación retinal.
Esta característica nos hace valorar otros posibles diagnósticos diferenciales pero dadas las características del cuadro clínico del ojo izquierdo se decide inyectar en la cavidad vítrea 0,025 mL/0,625 mg de bevacizumab (Avastin®, Roche, Basilea, Suiza). Este procedimiento se realizó sin complicaciones el 2 de febrero de 2021.
En la revisión de 24 horas no se observaron complicaciones derivadas del procedimiento y siete días después se percibió una disminución de los neovasos y la congestión, hemorragias en reabsorción. Los vasos continuaron solo en zona 1 y mantiene los exudados. En evaluaciones siguientes se constata una lenta mejoría por la reabsorción de las hemorragias, disminución del tamaño de los exudados y desaparición de los neovasos, pero sin evidencia de crecimiento vascular (Fig. 3).

Fig. 3 Imagen de fondo ojo en región temporal ojo izquierdo. Aplanamiento de los vasos, hemorragias en reabsorción y disminución del tamaño de los exudados.
A su vez el ojo derecho mantiene la línea demarcatoria que va alejándose, pasando a zona 3. Al mes de colocar la inyección intravítrea de anti-angiogénicos se realiza una fotocoagulación con láser para lo cual se emplea una consola de 810 nm (DC-3300, Nidek®, Gamagori, Japan) la periferia de la retina avascular del ojo izquierdo y también del ojo derecho. Para entonces su edad corregida era de 41,3 semanas y el peso 2 150 g.
En las semanas siguientes mantuvo la tendencia a la inactivación del ojo izquierdo. A las 47 semanas no presentaba hemorragias, ni neovasos, solo la presencia de exudados en las arcadas vasculares superior e inferior ojo izquierdo y era apreciable el buen efecto del láser en ambos ojos.
Discusión
La retinopatía de la prematuridad es un diagnóstico generalmente bilateral, y en ocasiones pueden detectarse distintos estadíos en ambos ojos, pero no encontramos reportes de cuadros tan asimétricos como el que presentamos, donde el ojo derecho cursa como una retinopatía de la prematuridad en estadío 1 en zona 2-3, con tendencia a la regresión espontánea y el ojo izquierdo se comporta como una retinopatía en zona 1, que nos plantea el posible diagnóstico de retinopatía de la prematuridad agresiva posterior.
Además de existir una asimetría en la agresividad, también se evidencia en la maduración vascular retinal pues en el ojo derecho estos crecieron hasta zona 3 y en el ojo izquierdo no superaron la zona 1.
Otro elemento que aporta atipicidad al cuadro es la presencia de exudados lipídicos. Los mismos están descritos en estadíos avanzados de retinopatía de la prematuridad (fases 4 y 5), que no coincide con este paciente.
Ante estas condiciones de atipicidad decidimos reevaluar el diagnóstico, considerando sus posibles diferenciales. No se excluye la presencia de otra enfermedad vascular en un paciente que al nacer de forma prematura y con factores de riesgo evidentes, pueda desarrollar retinopatía de la prematuridad.
Al tratarse de un paciente masculino sin lesiones en piel descartamos la incontinencia pigmenti (síndrome de Bloch-Sulzberger). Esta es una enfermedad muy rara, casi exclusivamente en mujeres debido a que se trata debe a una alteración genética de tipo dominante ligada al cromosoma X por lo que la mayoría de los varones mueren in útero, y los hallazgos en la piel generalmente son los primeros en aparecer. En el ámbito oftalmológico, desde el primer mes de vida se describen vasos de la retina tortuosos con ausencia de perfusión retinal periférica, hemorragias retinales en la unión de la retina vascular con la no vascular además de la posible presencia de otras anomalías como son catarata, estrabismo al nacer, nistagmo, así como la posible afectación del sistema nervioso central mediante ataques convulsivos, parálisis espástica y en edades más avanzadas puede ser evidente el retraso mental así como la aparición de otras alteraciones oftalmológicas como el desprendimiento de retina.7,8,9,10
Otro de los diagnósticos a valorar es la Enfermedad de Norrie, que es también una enfermedad infrecuente y, a diferencia de la anterior, tiene lugar en pacientes del sexo masculino por tratarse de un trastorno genético con patrón de transmisión recesivo ligada al cromosoma X. Su evolución es casi invariablemente hacia la ceguera por desprendimiento de retina en los primeros meses de vida, y aunque puede ser asimétrico no corresponde con el comportamiento del ojo derecho en este paciente, mucho más benigno.11,12
La enfermedad de Coats, si bien afecta a varones y es unilateral en el 90 % de los pacientes, no suele diagnosticarse a tan temprana edad y aunque es lógico pensar que en este caso su evaluación fue motivada por la condición de prematuridad, no por la presencia de síntomas como es habitual, tampoco apoya el diagnóstico la ausencia de telangiectasias, las dilataciones aneurismáticas de los vasos retinianos, el envainamiento vascular y, ocasionalmente neovascularización retinal 13. Si bien nuestro paciente presentaba exudados intra y subretinianos, el diagnóstico no es sustentable por la ausencia de los elementos clínicos primeramente expuestos.
Otra enfermedad, que a nuestro juicio podría estar en el diagnóstico diferencial de nuestro paciente por su similitud, es la vitreorretinopatía exudativa familiar, pues es una entidad en la que un fallo en la angiogénesis retiniana conduce a una vascularización incompleta en su periferia lo que origina isquemia y esta conlleva a la neovascularización retinal, exudación lipídica subretiniana y anormalidades en la interfase vitreorretiniana. Ambos ojos suelen afectarse, aunque de forma característicamente asimétrica.14
Años atrás, la historia clínica era crítica para distinguir la vitreorretinopatía exudativa familiar de la retinopatía de la prematuridad por la ausencia de prematuridad, bajo peso al nacer o la existencia de un tratamiento con oxígeno, así como la presencia de antecedentes familiares de la enfermedad. Se ha demostrado que esas condiciones no son suficientes porque en la vitreorretinopatía exudativa familiar el 50 % de los pacientes no tienen una mutación conocida, en algunos la enfermedad puede deberse a una nueva mutación; y hasta el 90 % de los familiares suelen ser asintomáticos, detectados solo cuando se realiza un examen clínico de la periferia retinal y/o hallazgos a la angiografía con fluoresceína. A esto se suma que los avances en la tecnología de la infertilidad y la atención neonatal han hecho que aumente la tasa de supervivencia de los recién nacidos, por lo tanto, se reconocen más casos de bebés prematuros con características de vitreorretinopatía exudativa familiar. Cuando se sospecha la presencia de vitreorretinopatía exudativa familiar en un paciente prematuro y clínicamente se observan características de ambas entidades se utiliza el término ROPER/fROP para describirlo.19 Otros autores prefieren discriminar en dos tipos: vitreorretinopatía exudativa familiar prematuro, cuando priman las características de vitreorretinopatía exudativa familiar y retinopatía de la prematuridad combinados con vitreorretinopatía exudativa familiar, cuando el cuadro es mixto y aquellos con una enfermedad más grave los llaman ROPER.6
Los estudios genéticos pudieran esclarecer, pero desfortunadamente no siempre es posible realizarlos debido a su costo y además un resultado negativo no descarta el diagnóstico por lo anteriormente expresado. Los pacientes con mutación genética positiva tienden a tener fenotipos más graves que progresan y desarrollan desprendimiento de retina recurrente15 por lo que se sugiere, ante la duda de considerarse un paciente ROPER/fROP controlar más estrictamente su evolución y realizar precozmente la fotocoagulación de la retina avascular. El caso presentado tiene varios factores de riesgo para desarrollar retinopatía de la prematuridad: nacimiento pretérmino (31,1 semanas), 1 100 g de peso, sexo masculino, oxigenoterapia, transfusiones, sepsis. A su vez presenta características sugestivas de vitreorretinopatía exudativa familiar: asimetría marcada y exudados retinales. No hay antecedentes familiares referidos de enfermedades retinianas ni ceguera, por lo que se impone una revisión oftalmológica a los padres, incluyendo angiografía fluoresceínica retinal, lo cual ha sido pospuesto debido a las restricciones que impone la actual pandemia de COVID-19.
Después de realizado este análisis de las posibles entidades del paciente y teniendo en cuenta los aspectos pendientes como el estudio genético, examen de los padres y la evolución en el tiempo los autores consideramos no descartar dos posibilidades diagnósticas: la primera es que sea solo portador de una retinopatía de la prematuridad, cuyos factores de riesgo son evidentes, con algunos signos de atipicidad en el ojo izquierdo. La segunda es que se trate de un caso ROPER, es decir, un paciente con una mutación en los genes de la vía Wnt (ya sea heredada o de novo) al que se añadió la retinopatía del prematuro por su condición al nacer.

