










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Archivo Médico de Camagüey
On-line version ISSN 1025-0255
AMC vol.5 no.1 Camagüey Jan.-Feb. 2001
ARTÍCULOS ORIGINALES
Aspectos clínicos y genéticos de la fibrosis quística
Clinical and genetical aspects of the cystic fibrosis
Dra. Elisa Dyce Gordon; Ángel Fernández Padrón; Adela Avilés Álvarez
Hospital Pediátrico Provincial Docente Dr. Eduardo Agramonte Piña.Camagüey, Cuba.
RESUMEN
Con el objetivo de escribir ciertos aspectos clínicos y genéticos de la fibrosis quística, se realizó un estudio descriptivo con 35 pacientes fibroquísticos registrados por la comisión provincial que atiende esta enfermedad, desde 1977 hasta 1995, a través de la revisión de historias clínicas y entrevistas a los padres. Las manifestaciones respiratorias resultaron las más frecuentes. El 91.42% (N=32) de los pacientes iniciaron la enfermedad durante la lactancia y de ellos 17 están ya fallecidos. La mayor mortalidad se presentó en aquellos menores de un año (N=13; 76.48%). El 25% de los cromosomas estudiados portaban la mutación mayor (AF508) y la presencia de ésta, incluso, en los heterocigotos se asoció a los cuadros clínicos más severos que llevaron a los niños más afectados a la muerte antes de los cinco años de vida. El 74.19% (N=23) de las parejas de riesgo recibieron asesoramiento genético. De ellos, cinco han solicitado diagnóstico prenatal en un posterior embarazo de uno de ellos. En conclusión, la fibrosis quística sigue siendo fatal en nuestro medio, por lo que se requiere del diagnóstico y tratamiento precoz, así como perfeccionar el asesoramiento genético y la caracterización molecular de las familias, con el fin de prevenirla .
DeCS: FIBROSIS QUISTICA; GENETICA; SINTOMAS CLINICOS.
ABSTRACT
To describe some clinical and genetic aspects of Cystic Fibrosis, a descriptive study on 35 fibrocystic patients registered by the Cystic Fibrosis Provincial comision since 1977 to 1995, througth clinical records review and parents interview was carried out. Respiratory symptoms were the most frequent 91.42% (N=32) of patients, initiated the disease during the firts year of life, and 17 of them are already deceased. Most of the patients disd during the lactation period 25% of all the studied chromosomes carried de major mutation (A F 508) and the presence of this mutation, ever in the heterocygotes was associated with the most severe clinical manifestations that lead the affected children to death before five years 74.19% of couples risk received genetic counselling. Five out of them subsequent pregnancy termination of one of them. In inclusion, Cystic Fibrosis continues being fatal in our milietr, that is why early diagnosis and treatment are necessary, as well as genetic couselling must be improved and molecular characterization of fibrocystic families must be don with aim of preventing the disease.
DeCS: CYSTIC FIBROSIS; GENETICS; SYMPTOMS CLINICAL.
INTRODUCCIÓN
La fibrosis quística (FQ) es una enfermedad generalizada, genéticamente determinada que afecta en Cuba alrededor de uno por cada 5 000 nacidos.1,2 Afecta funciones muy importantes del individuo. Sus manifestaciones clínicas se deben a anomalías funcionales en el epitelio de las vías aéreas pulmonares, del páncreas, del tracto gastrointestinal y glándulas sudoríparas fundamentalmente. Son las obstrucciones pulmonares y las infecciones, las manifestaciones que en mayor riesgo ponen la vida del paciente.
Estos enfermos y sus padres sufren mucho; los primeros porque les cuesta mucho trabajo respirar, porque tienen sentimiento de rabia e impotencia por no poder alcanzar ciertas metas que se han trazado en sus vidas, verguenza, baja autoestima y miedo a la muerte, entre otras.3 Y los segundos, porque tienen sentimiento de culpa, sufren por luto anticipado y tienen miedo a una futura gestación (Gaidzinski D; Giuliani R. Fibrose Cística: Estudio sobre o impacto de uma doenca genética crónica do núcleo familiar. I Congreso Latinoamericano de Pesquisaje Neonatal y Enfermedades Heredometabólicas. Libro de resúmenes. La Habana : Hotel Tritón, 14-18 sept. 1997.P 63-4).
Hasta el momento no existe tratamiento curativo para esta enfermedad. En los últimos años la esperanza de vida para estos enfermos ha aumentado notablemente, principalmente en países desarrollados.4 Sin embargo, en Cuba, específicamente en Camagüey, la FQ continúa siendo una seria y fatal enfermedad. 5 Con el objetivo de describir los aspectos clínicos y géneticos de la FQ en la población camagüeyana se realizó este trabajo, con el cual se contribuirá a perfeccionar la atención a estos pacientes, así como a la prevención de la enfermedad.
MÉTODO
Se realizó un estudio descriptivo con 35 pacientes fibroquísticos registrados en la Comisión provincial de fibrosis quística, nacidos entre 1977 y 1996 en el Hospital Pediátrico Provincial Docente Dr. Eduardo Agramonte Piña de Camagüey, con el objetivo de describir aspectos clínicos y genéticos importantes de la enfermedad.
Se revisaron todas las historias clínicas, de donde se extrajeron los datos necesarios para la investigación, tales como: nombres y apellidos del paciente, dirección particular, edad de inicio de la enfermedad, manifestaciones clínicas y evolución.
Además, los padres fueron entrevistados para saber si habían recibido asesoramiento genético (AG) o no, confeccionar el árbol genealógico y obtener su autorización para la realización de los estudios moleculares. Estos últimos fueron realizados en el laboratorio de genética molecular del Centro Nacional de Genética Médica por técnicas de amplificación molecular (PCR), en busca de la mutación mayor del gen (F 508) u otras estudiadas en nuestro país.
Los diagnósticos prenatales (DP) fueron realizados por dos procedimientos: uno directo (molecular) en las familias caracterizadas molecularmente y otro indirecto, a través de la cuatificación de las enzimas pancreáticas del feto presentes en el líquido amniótico, cuando la familia no estaba molecularmente estudiada.
Los estudios obtenidos fueron procesados mediante el paquete estadístico SPSS en los cuales se realizaron distribuciones de frecuencia a las variables categóricas: manifestaciones clínicas, resultados de los estudios moleculares y AG, asicomo a la variable númerica, edad, a la cual se le realizó la estadística descriptiva. Además se realizó prueba de hipótesis de proporciones a dicha variable con un grado de confiabilidad del 95% y nivel de significación de P< 0,05.
Los resultados se presentaron en forma de tablas y gráficos.
RESULTADOS
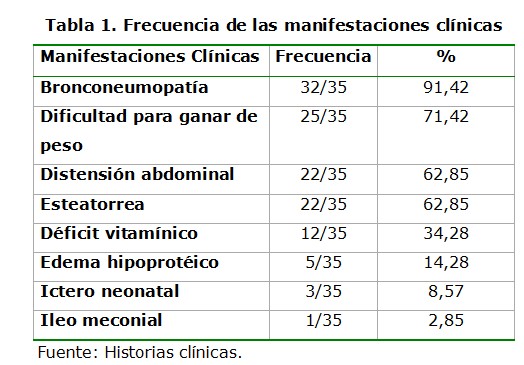
Las manifestaciones clínicas del aparato respiratorio fueron las más frecuentes; seguidas por la dificultad para ganar de peso, la distensión abdominal y la esteatorrea (tabla 1).
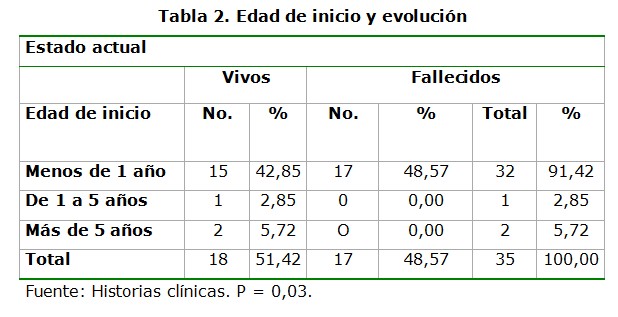
El inicio precoz de la enfermedad (durante la lactancia) fue el predominante (91,42%). El 48.57% de los pacientes que iniciaron la enfermedad con menos de un año de edad fallecieron. Es de notar que la proporción de fallecidos que comenzaron su enfermedad con menos de un año es significativamente mayor que la proporción de los que se mantienen vivos, que comenzaron también su enfermedad con menos de un año (p= 0,03) (tabla 2).
El 76, 48% de los pacientes (N=13) fallecieron durante el primer año de vida (tabla 3)
La edad media al fallecer fue de 10,2 meses.
De los 18 pacientes molecularmente caracterizados, presentaron la mutación mayor ? F508, solo seis. No se encontraron otras mutaciones. La mutación mayor se presentó en doble dosis (homocigoto) en tres pacientes y en dosis simple (heterocigoto) en otros tres pacientes, es decir, esta mutación apareció en nueve de los 36 cromosomas fibroquísticos estudiados (25%). Es de destacar, que tanto los pacientes homocigotos como los heterocigotos para esta mutación se encuentran ya fallecidos, cuatro durante el primer año de vida y dos entre uno y cinco años. Los pacientes que no presentaban esta mutación están todos vivos.
Los 35 pacientes estudiados estaban distribuidos en 31 familias (27 con un solo hijo afectado y cuatro con dos hijos afectados). Del total de familias, recibieron AG 23 (74.19%).
De estas parejas de riesgo, algunas se han separado, otras han renunciado tener descendencia, algunos están indecisos y hasta el momento cinco parejas han acudido a consulta solicitando DP. El diagnóstico prenatal fue realizado por técnicas enzimáticas en dos parejas y por estudios moleculares en tres.
Se obtuvieron tres resultados normales y dos patológicos (uno enzimático y otro molecular). La pareja que obtuvo el resultado molecular patológico (feto ? F508/ ?F508) optó por continuar con el embarazo, naciendo como producto del mismo una niña fibroquística que falleció a los seis meses de edad.
DISCUSIÓN
En América Latina la edad media al fallecimiento de los pacientes con FQ es de alrededor de 8,4 años. (Macri CN Fibrosis Quística (mucoviscidosis): clínica de las manifestaciones broncopulmonares. VIII Congreso Latinoamericano y III Jornada Hispanoamericana de Fibrosis Quística. Libro de resúmenes . La Habana: Palacio de las Convenciones, 20-23 de Octubre 1997: 62-64). En el presente trabajo resultó ser de 10,2 meses, debido en parte a que la mayor mortalidad se produjo previo y al inicio de la creación de la comisión provincial.
Casi el 50% de los pacientes estudiados fallecieron y todos iniciaron la enfermedad durante la lactancia, lo cual pudiera estar relacionado en parte al desarrollo incompleto de las vías respiratorias, inmadurez del sistema inmunológico durante la lactancia, o ambos, presente en estos niños, e incluso en niños normales.6
Por otra parte, la enfermedad presenta considerable variación en cuanto a severidad entre los diferentes pacientes.7 Esta heterogeneidad clínica es atribuible a las diferentes mutaciones específicas del gen de la FQ presentes en los pacientes6,8 ya descritas en más de 650 y donde la más frecuente es la ? F508.1,4,6,8
Aunque el genotipo no explica totalmente las variaciones clínicas entre los pacientes2,7 es conocido que la mutación ?F508 es respobnsable de las formas clínicas más severas (aquellas acompañadas de insuficiencia pancreática).1 En los pacientes homocigóticos (? F508 / ? F508) aparecen los primeros síntomas antes de arribar a los seis meses de edad, tienen una función pulmonar más pobre, la incidencia de infecciones por pseudomona aeruginosa y mortalidad temprana es mayor que en los pacientes heterocigotos,9 acorde con los resultados expuestos. También es conocido que heterocigotos compuestos G 551D/ ? F508 son clínicamente indistinguibles de los homocigotos para la mutación ? F50810 y que heterocigotos ? F508 llegan a la adultez posiblemente portando una mutación que se expresa clínicamente más leve en el otro cromosoma.8
Estableciendo una relación entre los conocimientos, los resultados obtenidos, así como teniendo en cuenta que en nuestro país las únicas mutaciones estudiadas son la ? F 508, G542X, R1162X y N1303K sería lógico pensar:
1 Que la presencia de al menos un alelo ? F 508 se relaciona con el desarrollo de síntomas más tempranos de la enfermedad y por lo tanto, con el peor pronóstico.
2. Que en nuestros pacientes heterocigotos compuestos (? F508/ otra mutación), esta otra mutación no estudiada en nuestro medio esté también relacionada con cuadros clínicos severos.
No obstante, no pueden olvidarse otros factores que pueden modificar las manifestaciones clínicas y pronóstico de la enfermedad, tales como el efecto modificante de genes en otros loci no FQ y el ambiente,10 así como el diagnóstico y tratamiento temprano.2 La menor frecuencia de presentación de la mutación mayor (? F 508) en la población estudiada (25%) con respecto al 34% encontrado en el país (Collazo T, Granda H: Rojo M. Caracterización molecular y estudio de los polimorfismos en 220 pacientes fibroquísticos en Cuba VIII Congreso latinoamericano y III Jornada hispanoamericana de Fibrosis Quística. Libro de resúmenes. La Habana Palacio de las Convenciones, 20-23 de Octubre 1997.p 62-4).
CONCLUSIÓN
La FQ se mantiene siendo una enfermedad incurable y fatal para un gran número de pacientes. Esta mortalidad está relacionada con la edad de inicio de la enfermedad, así como por la presencia de la mutación ? F508 en los cromosomas fibroquísticos. Por lo tanto, para estos pacientes es de gran importancia el diagnóstico y tratamiento precoz. Se le debe ofrecer siempre la posibilidad de realización de estudio molecular a todos los niños con FQ y caracterizar molecularmente a la familia, ya que el resultado de este estudio puede predecir el curso clínico de la enfermedad, y es muy útil además para ofrecer AG y DP a las parejas de riesgo.
REFERENCIAS BIBLIOGRÁFICAS
1. Gispert S. Identificado el gen de la Fibrosis Quística. Resumed. 1991;4(1):3-7.
2. Rojo M. Hablemos de fibrosis quística. Av Med Cub. 1997;4(12):35-7.
3. Ibáñez MT. Sintomatología física y factores psicosociales en fibrosis quística. Fibr quíst.1996;15:5.
4. Asencio O, Bosque M, Marco MT. Fibrosis quística: Una nueva patología del adulto. An Esp Pediatr.1997;46(1):24-28.
5. Dyce E, Llevara Y, Avilés A, Guerrero MA. Aspectos clínicos, histopatológicos y genéticos de la fibrosis quística. Arch Med Cam. 1994;1(1):8.
6. Fernández MR, Rubio J. Factores predisponentes de infecciones respiratorias agudas en el niño. Rev Cub Med Gen Int. 1993;6(10):400-8.
7. Scriver CR, Fujiwara TM. Invited editorial: Cystic Fibrosis genotypes and views on screening are both heterogenous and population related. Am J Hum Genet. 1992;51:943-50.
8. Beaudet AL. Genetic testing for Cystic Fibrosis. Medical Genetics. 1992;39(2):213-28.
9. Johansen HK, Nir M, Hoiby N, Kovh C, Schwartz M. Severity Cystic Fibrosis in patients homozygous and heterozygous for delta ? F 508 mutation. Lancet. 1991;337(8742):631-4.
10. Hamosh A, King TM, Rosenstein B, Corey M, Levison H, Durie P. Cystic Fibrosis patients bearing both the common missense mutations gly-aspbat codon 551 and the ? F508 mutation are clinically indistinguishable from ? F508 homozygotes, except for decreased risk of meconium ileus. Am J Hum Genet. 1992;51:245-50.
Recibido. 3 de octubre de 2000
Aprobado:15 de diciembre 2000
Dra. Elisa Dyce Gordon Especialista de II Grado en Genética Clínica. Profesor Asistente. Hospital Pediátrico Provincial Docente Dr. Eduardo Agramonte Piña


{kind=link}
{kind=link}
{kind=link}