










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
En la producción de biológicos, especialmente en aquellos obtenidos a partir de microorganismos, resulta imprescindible contar con un suministro estable de células, como punto de partida para la producción. El concepto de Sistema de Lotes de Siembra en niveles, en el cual el Lote de Siembra de Referencia (LSR) es usado para generar el Lote de Siembra de Trabajo (LST), es considerado el enfoque más práctico para lograrlo.1 Estas producciones parten de un sistema de lotes semillas, para prevenir los cambios no deseados como mutaciones espontáneas, así como de las propiedades bioquímicas que se producen con subcultivos repetidos o generaciones múltiples. El sistema de lotes semillas consiste en fabricar lotes sucesivos de un producto mediante cultivos de células derivadas de un mismo aislamiento original.2 A partir de un aislamiento original se constituye el Lote Maestro (LM), que da lugar al LSR, del cual posteriormente se elabora el LST. Con ellos, se inicia el proceso de producción. El proceso de elaboración y el método de conservación de cada LST, una vez establecido, debe ser el mismo para minimizar los cambios que pudieran producirse en el proceso productivo.
Los procesos de obtención de los LST para fabricar los Ingredientes Farmacéuticos Activos (IFA) en las plantas de producción del Instituto Finlay de Vacunas (IFV), presentan una secuencia de pasos similares: atemperamiento del criotubo del LSR, homogenización, siembra en medio de cultivo, incubación en condiciones controladas específicas para cada microorganismo, determinación de parámetros críticos, pureza del cultivo, identificación, control de calidad, conservación y almacenamiento.3,4,5
Al agotarse el LST en uso, este debe ser sustituido por un nuevo LST fabricado a partir del mismo LSR que el LST agotado.1 La sustitución del LST constituye un cambio y debe ser documentado. También deben valorarse los riesgos que implica este cambio.6) El procedimiento asegura que los cultivos de producción mantengan las mismas características de la cepa de origen para garantizar la consistencia de expresión de los antígenos de interés. Este sistema garantiza un número de pases fijos entre el lote de referencia y los cultivos de producción.7
Según la Regulación 33/2003 del Centro para el Control Estatal de Medicamentos, Equipos y Dispositivos Médicos (CECMED), el concepto del cambio se refiere a cualquier modificación introducida en el proceso de producción o el control, que conlleva a variaciones en aspectos contenidos en el Registro Sanitario de Medicamentos. Se muestra como ejemplo representativo de cambio menor, el establecimiento de un nuevo Banco de Células de Trabajo derivado de un Banco Maestro de Células, previamente aprobado.6,8,9
El IFV cuenta con alta experiencia y una plataforma tecnológica industrial para la obtención de vacunas a partir de IFA. Estas son de diferentes tipos: polisacarídicas, a partir de anatoxinas, de suspensión de células inactivadas y de tipo conjugada.7 Las mismas se obtienen en las siguientes instalaciones productivas del IFV: Ingrediente Farmacéutico Activo 1 (IFA1), Difteria, Pertussis y Tétanos (DPT), Ingrediente Farmacéutico Activo 2 (IFA 2), Planta Multipropósito de Síntesis Química (PMSQ).
Para mantener las producciones biofarmacéuticas a un nivel competitivo, se hace imprescindible la aplicación de un enfoque de riesgos sobre la calidad del producto concebido sobre la base del conocimiento científico y que proporcione la seguridad necesaria destinada a los pacientes dentro del propio ambiente regulatorio de estándares prestablecidos.8,10 Como parte de la Gestión de Riesgo a la Calidad (GRC), la evaluación del riesgo es el proceso global de identificación, análisis, valoración y revisión del riesgo.9,11 Por último, se realiza una revisión sistemática a través de seguimientos programados, evaluaciones o auditorías.
Dentro de las técnicas y herramientas empleadas en la GRC están: diagrama de flujo, método 6M, diagrama causa-efecto, Análisis Modal de Fallos y Efectos (AMFE), Análisis Modal de Fallos, Efectos y su Criticidad, Análisis por Árbol de Fallos, Análisis de Peligros y Puntos Críticos de Control y Análisis Preliminar de Peligros. La herramienta a aplicar está en dependencia de la complejidad del proceso.10,12
Evaluar la gestión de riesgos en un proceso de cambio en los LST utilizados en las producciones de vacunas del IFV constituye el objetivo del trabajo. Para ello, se basó en una técnica empleada en el análisis de riesgos AMFE con el propósito de identificar, analizar y evaluar los riesgos, determinar el impacto en la calidad de las vacunas y proponer una estrategia a partir de las acciones derivadas.
Materiales y Métodos
Microorganismos incluidos en el análisis de riesgos
Los microorganismos incluidos en el análisis de riesgo se muestran en la Tabla 1.
Tabla 1 Microorganismos incluidos en el análisis de riesgo.
| Especie | Serotipos |
|---|---|
| serogrupo A | |
| serogrupo B | |
| serogrupo C | |
| serogrupo C | |
| serogrupo W135 | |
| serotipo 1 | |
| serotipo 5 | |
| serotipo 6B | |
| serotipo 14 | |
| serotipo 18C | |
| serotipo 19A | |
| serotipo 23F | |
Causas de la sustitución de los LST
En la producción de un IFA, el cambio de LST debe ocurrir con la menor frecuencia posible, ya que el LST constituye la materia prima de partida fundamental del proceso y la fuente importante de variaciones en el producto obtenido. Este debe ser sustituido por agotamiento, resultados fuera de especificación (RFE) del LST o cambios en el comportamiento de los resultados de los procesos. Los RFE en los procesos de producción pudieran estar vinculados o no al LST, pero entre las decisiones tomadas en producción está la sustitución del LST. Otra causa de cambio de LST es la ocurrencia de eventos que pudieran poner en riesgo los resultados del proceso, aunque no se hayan producido RFE, como pudieran ser el aumento de la temperatura durante el almacenamiento, posible mezcla de lotes, entre otras.
El análisis de riesgo que se muestra en el presente artículo no es aplicable si la causa del cambio es por RFE del LST, o en los casos donde el RFE de los lotes de procesos de IFA estén provocados por el LST. En esos casos debieran ser analizadas las causas de los RFE para incluir otros riesgos, aparte de los mostrados. Tampoco es aplicable al cambio de LSR, aunque los riesgos y su evaluación pudieran ser similares.
Condiciones del entorno
La aplicabilidad de este análisis está determinada por las siguientes condiciones del entorno: el nuevo LST es fabricado a partir del mismo LSR, en las mismas instalaciones y empleando igual metodología (proceso de fabricación, método y temperatura de conservación, tamaño del lote, etc.) que el LST anterior. El nuevo LST es fabricado con materias primas aprobadas por el sistema de calidad del IFV, dentro del periodo de vigencia establecido por el fabricante y de proveedores aprobados por el IFV.
Aplicación de la Gestión de Riesgos a la Calidad
Se conformó un equipo de trabajo integrado por varios especialistas para lograr un enfoque multidisciplinario en la definición del problema y el alcance del mismo; así como establecer los objetivos y consideraciones e identificar los riesgos potenciales que pudieran influir en un posible cambio de LST. Los especialistas procedieron de diferentes áreas del IFV (Producción, Control de la Calidad y Aseguramiento de Calidad), con experiencia en la actividad en que se desempeñaban y formación en microbiología, bioquímica, ingeniería química y farmacia, fundamentalmente.
Metodología empleada
En la ejecución del análisis de riesgo se aplicó el AMFE, según la metodología descrita en el Procedimiento Normalizado de Operación (PNO) establecido por el Sistema de Gestión de la Calidad del IFV. Para la identificación de los riesgos se emplearon diferentes técnicas como la tormenta de ideas, los cinco por qué y las seis M. Otra herramienta cualitativa que fue utilizada fue el método Delphi, el cual permite obtener un consenso fiable de la opinión de un grupo de expertos.13 Posteriormente, se determinaron las causas de los efectos identificados a través del método de Ishikawa o espina de pescado.14
El AMFE es un procedimiento de análisis de fallos potenciales que clasifica estos según la gravedad o por el efecto de los mismos en el sistema.12 Se analizó el tipo y probabilidad de ocurrencia de cada una de las fallas en el cambio de LST y la evaluación de su impacto en la calidad del producto final. Estos criterios se reflejaron en una matriz de riesgo que se muestra en la Tabla 2. Los valores de riesgo (VR) pueden variar desde 1 punto (valor mínimo) hasta 25 puntos (valor máximo). La escala de los criterios del grado de impacto o severidad (I) y probabilidad de ocurrencia (P) son totalmente arbitrarios, de este modo la selección ocurre según la experiencia del investigador.
Tabla 2 Matriz de riesgo para la evaluación de la criticidad del riesgo
| Impacto | ||||
|---|---|---|---|---|
| Estimación del valor de riesgo (VR) | Bajo (1 punto) | Medio (3 puntos) | Alto (5 puntos) | |
| Probabilidad | Baja (1 punto) | 1 | 3 | 5 |
| Media (3 puntos) | 3 | 9 | 15 | |
| Alta (5 puntos) | 5 | 15 | 25 | |
Por tanto, se definieron tres rangos de evaluación para encauzar las decisiones sobre el tipo de acción a tomar:
Probabilidad (P): frecuencia con que se presenta el riesgo. Alta (5 puntos): es muy probable que el riesgo se presente y ocurra siempre. Media (3 puntos): es probable que el riesgo se presente y suceda en algunas ocasiones. Baja (1 punto): es muy poco probable que el riesgo se presente y ocurre raras veces.
Impacto (I): forma en la cual el riesgo podría afectar los resultados del proceso. Alto (5 puntos): afecta en alto grado el proceso. Medio (3 puntos): afecta en grado medio al proceso. Bajo (1 punto): afecta en bajo grado al proceso.
A partir de esa evaluación se estimó el VR o prioridad para cada fallo, donde: VR= P x I. Si VR > 9, entonces el riesgo es crítico, por lo tanto, existe una alta prioridad (A) para investigarlo y accionar sobre él inmediatamente. Si 3 ≤ VR ≤ 9, entonces el riesgo es mayor, por lo tanto, existe una prioridad media (B) para accionar sobre él. Si VR < 3, entonces el riesgo es menor, por lo tanto, existe una prioridad baja o insignificante (C).
Con los riesgos priorizados, se identificó la existencia de controles asociados y su efectividad, con el fin de determinar el nivel de riesgo. Se utilizó la clasificación siguiente: no existen controles (1 punto), existen controles y no son efectivos (2 puntos), existen controles y no están documentados (3 puntos), los controles están documentados y son efectivos (4 puntos).
Posteriormente el nivel de riesgo fue clasificado de la forma siguiente:
Extremo: riesgos con priorización alta (A) y media (B), sin controles (1 punto). Riesgo no aceptable.
Alto: riesgos con priorización alta (A) y media (B), y controles no efectivos (2 puntos). Riesgo no aceptable.
Moderado: riesgos con priorización alta (A) y media (B), con controles efectivos, pero no documentados (3 puntos). Riesgo aceptable.
Bajo: riesgos con priorización alta (A) y media (B), con controles documentados y efectivos (4 puntos). Riesgos con priorización baja (C). Riesgo aceptable.
Con el nivel de prioridad y la identificación de la existencia de controles se clasificó a cada modo de fallo con el nivel de riesgo necesario y según su nivel de aceptación. A partir de estos resultados se propuso una estrategia con vistas a minimizar los riesgos. Se utilizó Microsoft Excel como herramienta para facilitar la ponderación de los datos, evaluación y control.
Como parte del Sistema de Gestión de Riesgo, estos resultados se comunican a las partes interesadas y se realiza una revisión sistemática a través de seguimientos programados, evaluaciones o auditorías.
Resultados y Discusión
Riesgos potenciales
Los riesgos identificados se muestran en la Tabla 3.
Tabla 3 Riesgos identificados.
| Riesgo | Descripción |
|---|---|
| R1 | Inhibición del crecimiento durante la fabricación del LST |
| R2 | Contaminación del LST |
| R3 | Resultados fuera de especificación durante los controles de calidad del LST |
| R4 | Inhibición del crecimiento celular en los procesos realizados a partir del LST |
| R5 | Contaminación de los procesos realizados a partir del LST |
| R6 | No expresión del IFA en los procesos realizados a partir del LST |
| R7 | Aumento de los contaminantes en los lotes de IFA fabricados a partir del LST |
| R8 | Dificultades en la estabilidad del LST |
Probables causas de ocurrencia de los riesgos potenciales identificados
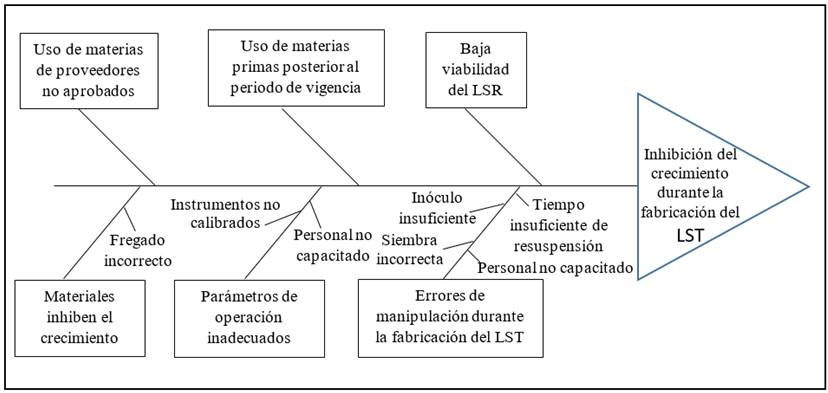
El grupo de trabajo identificó las probables causas de los riesgos potenciales identificados, un resumen de las cuales se muestra en la Tabla 4. Dada la complejidad de los riesgos R1 y R2 se empleó el diagrama de Ishikawa (Fig. 1 y 2) para evaluar la probabilidad de ocurrencia de los mismos.
R1: Inhibición del crecimiento durante la fabricación del LST
Durante el proceso de fabricación del LST puede haber inhibición del crecimiento, provocado por la baja viabilidad del LSR. Otras posibles causas de inhibición podrían ser el uso de materias primas de proveedores no evaluados o uso posterior al período de vigencia establecido por el fabricante. Estas últimas dos causas no son objetivos del presente análisis. En análisis similares planteados por otros autores,10,12 en caso de que alguna materia prima deba ser utilizada en esas condiciones, debe realizarse un análisis de riesgo antes de su uso, para identificar el impacto sobre el proceso.
Los errores durante la manipulación del cultivo y parámetros de operación inadecuados pudieran ser causa de inhibición del crecimiento. Entre estas fallas están no esperar el tiempo suficiente para la resuspensión de la pastilla liofilizada, no usar el suficiente inóculo, siembra o inoculación incorrecta y no cultivar en las condiciones (temperatura, agitación, aireación, anaerobiosis) requeridas. Estos errores son causados por trabajar con personal no capacitado y utilizar instrumentos no calibrados. El uso de materiales inadecuados (fregado incorrecto, materiales de proveedores no evaluados) pudiera inhibir el crecimiento, según referencias de la literatura.8,15,16
R2: Contaminación del LST
Durante el proceso de fabricación del LST pudiera ocurrir contaminación, originada por el uso del LSR, medios de cultivo, soluciones o materiales contaminados.1,16 Estos pudieran estar contaminados porque su esterilización o conservación no fue adecuada. El uso de materiales desechables fuera del período de vigencia o proveedores no evaluados pudiera ser fuente de contaminación.
Otra posible causa de contaminación es que en el Gabinete de Seguridad Biológica (GSB) no se garantice la pureza del cultivo, debido a errores de manipulación, rotura del filtro HEPA o funcionamiento inadecuado del equipo.13
La manipulación incorrecta en cualquier etapa del proceso puede provocar contaminación del LST. Estos errores son causados por trabajar con personal no capacitado.
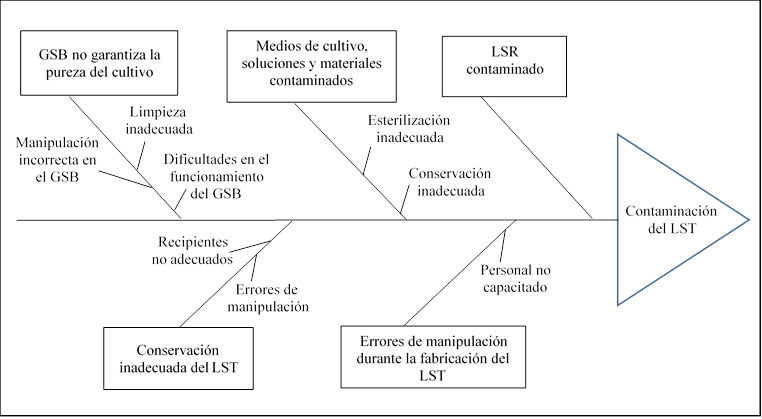
La conservación inadecuada del LST pudiera ser otra causa de contaminación, debido a viales mal cerrados por manipulación incorrecta o tapas inseguras asociadas a proveedores no calificados.
R3: Resultados fuera de especificación durante los controles de calidad del LST
Este riesgo está más relacionado con el LSR y no con el proceso de fabricación. Pudiera ser baja viabilidad (R3.1), contaminación (R3.2) e incumplimiento de otras especificaciones de calidad (R3.3).
R4: Inhibición del crecimiento celular en los procesos realizados a partir del LST
Las causas para este riesgo son las mismas que para el R1, aunque refiriéndose al LST y no al LSR. En este caso se adiciona una nueva causa: la conservación y/o manipulación incorrecta del LST.
R5: Contaminación de los procesos realizados a partir del LST
Las causas para este riesgo son las mismas que para el R2, aunque refiriéndose al LST y no al LSR. Además, los procesos pudieran contaminarse en pasos posteriores como la fermentación. La causa de la contaminación en estos pasos pudiera ser funcionamiento inadecuado del fermentador. Esta causa no está vinculada al LST.
R6: No expresión del IFA en los procesos realizados a partir del LST
El envejecimiento de un LST pudiera influir en la no expresión de los elementos que constituyen un IFA, ya sea por no expresión de genes que codifican a proteínas, expresión de polisacáridos en forma y composición o algunas mutaciones espontáneas. Cuando se produce el cambio del LST esta condición no existe.17 Las causas de este riesgo no están vinculadas al cambio del LST.
R7: Aumento de los contaminantes en los lotes de IFA fabricados a partir del LST
Generalmente, el aumento de los contaminantes se produce por las condiciones de la fermentación y la purificación.7,16,17 Las causas de este riesgo no están vinculadas al cambio del LST.
R8: Dificultades en la estabilidad del LST
La estabilidad del LST puede afectarse si las condiciones de almacenamiento no son adecuadas, el medio y el método de conservación no es el idóneo o la concentración celular no es la apropiada.1
Tabla 4 Causas de los riesgos potenciales identificados.
| Riesgo | Causa |
|---|---|
| R1 | C1: Baja viabilidad del LSR |
| C2: Uso de materias primas posterior al periodo de vigencia | |
| C3: Uso de materias primas de proveedores no evaluados | |
| C4: Errores de manipulación | |
| C5: Parámetros de operación inadecuados | |
| C6: Uso de materiales no evaluados | |
| R2, R3.2 | C7: LSR contaminado |
| C8: Uso de medios de cultivo, soluciones y materiales contaminados | |
| C9: En el GSB no garantiza la pureza del cultivo | |
| C10: Errores de manipulación | |
| C11: Conservación inadecuada del LST | |
| R3.1 | C1: Baja viabilidad del LSR |
| C2: Uso de materias primas posterior al período de vigencia | |
| C3: Uso de materias primas de proveedores no evaluados | |
| C4: Errores de manipulación | |
| C5: Parámetros de operación inadecuados | |
| C6: Uso de materiales no evaluados | |
| R3.3 | C12: LSR no cumple con las especificaciones de calidad |
| R4 | C13: Baja viabilidad de los LST |
| C14: Disminución de la viabilidad del LST debido a condiciones de almacenamiento | |
| C15: C2, C3 | |
| C15: C4, C5, C6 | |
| R5 | C7: LST contaminado |
| C8: Uso de medios de cultivo, soluciones y materiales contaminados | |
| C9: En el GSB no garantiza la pureza del cultivo | |
| C10: Errores de manipulación | |
| C11: Conservación inadecuada del LST | |
| C16: Funcionamiento incorrecto de los fermentadores | |
| R6 | - |
| R7 | - |
| R8 | C17: Condiciones no adecuadas de almacenamiento |
Evaluación de la probabilidad de ocurrencia de los riesgos
Para la evaluación de la probabilidad de ocurrencia se consideró el comportamiento de los procesos en las plantas de producción entre los años 2015 y 2020. La incidencia de la contaminación y la inhibición fue inferior al 5%. Ninguno de los procesos no satisfactorios en este período fue rechazado por causas relacionadas con el LST. La ponderación de la probabilidad de ocurrencia de los riesgos se muestra en la Tabla 5.
Evaluación del impacto de los riesgos
La afectación de la calidad de los LST pudiera provocar el rechazo de los mismos o de los procesos, por consiguiente, este hecho no tendría implicaciones en la calidad del IFA y la vacuna. En todos los casos la afectación a la calidad de los LST provocaría pérdidas en la empresa, según varios autores,5,8,10 debido al aumento de rechazos, o provocaría el incumplimiento de planes de producción por la demora en la obtención de productos satisfactorios. La ponderación de la probabilidad de impacto de los riesgos se muestra en la Tabla 5.
Establecimiento de prioridades de los riesgos: valor del riesgo
A partir de la ponderación del impacto y la probabilidad de los riesgos se obtuvo el valor del riesgo (VR) que se muestra en la Tabla 5. Los riesgos identificados fueron clasificados como riesgos mayores con prioridad media de actuar sobre ellos o riesgos críticos con alta prioridad para actuar sobre ellos.
Tabla 5 Ponderación y clasificación de los riesgos.
| Riesgo | Causa | P | I | VR | Clasificación |
|---|---|---|---|---|---|
| R1 | C1, C4, C5, C6 | 1 | 5 | 5 | B: riesgo mayor |
| C2, C3 | 3 | 5 | 15 | A: riesgo crítico | |
| R2 | C7, C8, C9, C10, C11 | 1 | 5 | 5 | B: riesgo mayor |
| R3.1 | C1, C4, C5, C6 | 1 | 5 | 5 | B: riesgo mayor |
| C2, C3 | 3 | 5 | 15 | A: riesgo crítico | |
| R3.3 | C12 | 1 | 5 | 5 | B: riesgo mayor |
| R4 | C13, C14, C4, C5, C6 | 1 | 5 | 5 | B: riesgo mayor |
| C2, C3 | 3 | 5 | 15 | A: riesgo crítico | |
| R5 | C7, C8, C9, C10, C11, C16 | 1 | 5 | 5 | B: riesgo mayor |
| R6 | - | 1 | 3 | 3 | B: riesgo mayor |
| R7 | - | 1 | 3 | 3 | B: riesgo mayor |
| R8 | C17 | 1 | 5 | 5 | B: riesgo mayor |
P: Probabilidad. I: Impacto. VR: Valores de riesgo. _: No se hallaron posibles causas.
Estado de los controles
Durante el proceso de fabricación del LST se evalúa la pureza del cultivo por tinción de Gram.1) Los lotes son evaluados por Control de la Calidad determinándose la viabilidad, realizándosele ensayos de pureza, pruebas bioquímicas, fisiológicas y serológicas para confirmar identidad según cada especificación. Los LST y LSR se chequean anualmente para evaluar estabilidad. En la esterilización por calor húmedo se utilizan cargas calificadas.2 En la autoclave se colocan bioindicadores semanalmente para comprobar el adecuado funcionamiento del equipo. Los equipos de conservación de los LST están conectados a sistemas de control supervisor que registran la temperatura de forma continua.2,16) Existen controles, están documentados y son efectivos. Por tanto, la evaluación para todos los riesgos fue de 4.
Clasificación del nivel de riesgo
La clasificación y el estado de los controles determinaron el nivel de cada riesgo identificado. Los resultados se muestran en la Tabla 6. Los riesgos identificados tienen un nivel de riesgo bajo y se consideran riesgos aceptables.
Tabla 6 Nivel de riesgo de los riesgos identificados.
| Riesgo | Clasificación | Estado de los controles | Nivel de riesgo | Nivel de aceptación |
|---|---|---|---|---|
| R1 | B: Mayor | 4 | Bajo | Riesgo aceptable |
| A: Crítico | 4 | Bajo | Riesgo aceptable | |
| R2 | B: Mayor | 4 | Bajo | Riesgo aceptable |
| R3 | B: Mayor | 4 | Bajo | Riesgo aceptable |
| A: Crítico | 4 | Bajo | Riesgo aceptable | |
| R4 | B: Mayor | 4 | Bajo | Riesgo aceptable |
| A: Crítico | 4 | Bajo | Riesgo aceptable | |
| R5 | B: Mayor | 4 | Bajo | Riesgo aceptable |
| R6 | B: Mayor | 4 | Bajo | Riesgo aceptable |
| R7 | B: Mayor | 4 | Bajo | Riesgo aceptable |
| R8 | B: Mayor | 4 | Bajo | Riesgo aceptable |
Estrategia a seguir para los cambios de LST
La metodología para realizar cambios de LST es la siguiente:
Presentación de la propuesta de cambio a Aseguramiento de la Calidad. El cambio debe cumplir con las condiciones establecidas en el acápite Causas de la sustitución del LST. Si no se cumple con esas condiciones pudiera actuarse de la forma siguiente:
El LST es elaborado a partir de un nuevo LSR. El cambio de LSR constituye un cambio mayor y en su evaluación debe fabricarse un LST y procesos a partir de este.
El LST es fabricado en instalaciones diferentes a donde habitualmente se realiza. Debe incluirse la evaluación del cambio de instalación dentro del mismo cambio y se realizará un nuevo análisis de riesgo.
Si cambia la metodología de fabricación del LST no es aplicable este análisis de riesgo.
Se utilizan materias primas de nuevos proveedores. Debe seguirse lo establecido por la metodología de evaluación de proveedores.
Fabricación del nuevo LST.
Envío de muestras del nuevo LST a Control de la Calidad.
Ejecución de los ensayos establecidos en la especificación.
Fabricación de lotes de IFA con el nuevo LST. Se realizará cuando los resultados de los controles de calidad sean satisfactorios
Entrega del informe de evaluación de cambio. Puede continuar la producción, pero los IFA no serán aprobados hasta la evaluación del informe de cambio por Aseguramiento de la Calidad.
Evaluación del cambio por la comisión de cambio del IFV.
Aprobación de los lotes de IFA por Aseguramiento de la Calidad.
Realización de estudios de estabilidad con un lote de IFA. Se seleccionarán los primeros lotes producidos con el nuevo LST.
Conclusiones
Se identificaron ocho riesgos y de ellos se valoraron 17 causas. Los fallos identificados en la gestión de riesgos relativa al cambio de los LST para la fabricación de vacunas en el IFV fueron aceptables, y las afectaciones, en caso de ocurrir, están vinculadas al cumplimiento de las tareas del cambio de un nuevo LST, no a la seguridad y eficacia del IFA, ni de las vacunas. Como consecuencia, se propuso una estrategia que garantiza el cumplimiento y revisión de las tareas establecidas con el propósito de mitigar los posibles riesgos ante un cambio de LST para la fabricación de un producto biofarmacéutico.

