










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.26 no.1 La Habana Jan.-Mar. 2009
REVIEW
G Protein-coupled receptors as targets for drug design
Los receptores asociados a proteínas G como blancos en el diseño de fármacos
Ania de la Nuez Veulens, Rolando Rodríguez
División de Química Física, Centro de Ingeniería Genética y Biotecnología, CIGB Apartado Postal, 6162, Ciudad de La Habana, Cuba
ABSTRACT
G protein-coupled receptors (GPCRs) are the target for more than 50% of the drugs currently on the market, including about 25% of the 100 top-selling drugs. They are considered the most important molecules in the field of drug discovery and design today, mostly due to their role as receptors in many of the basic processes in the body, and because they are present in all tissues. Unfortunately, a structure-based rational design is very difficult for GPCRs; the structures available for modeling purposes are only for family A. Despite this fact, research has continued and progressed, using combined structure-based techniques. This review intends to summarize this work.
Keywords: GPCR, 7TM, receptor, virtual screening, docking, molecular modeling, drug design.
RESUMEN
Los receptores asociados a proteínas G (GPCRs), son el blanco de más del 50% de los medicamentos que se encuentran actualmente en el mercado, e incluyen cerca del 25% de la lista de las 100 medicinas más vendidas en el mundo, estas moléculas son consideradas hoy en día como las más importantes para el diseño de fármacos, fundamentalmente por su papel como receptores de la mayoría de los procesos básicos del organismo, además de estar presentes en todos los tejidos. Desafortunadamente el diseño racional basado en la estructura se hace muy difícil para las GPCRs, las estructuras que existen son solo de la familia. A pesar de esto, las investigaciones han continuado y progresado utilizando técnicas combinadas. Esta revisión trata de resumir este trabajo.
Palabras clave: GPCR, 7TM, evaluación virtual, acoplamiento molecular, modelación molecular, diseño de fármacos.
INTRODUCTION
G protein-coupled receptors (GPCRs) form the largest family of membrane proteins responsible for communication between the cell and the environment. These proteins recognize extracellular messengers and transduce the signal to the cytosol. GPCRs bind to a wide variety of molecules, including ions, amino acids, peptides, lipids, and nucleotides. They control the activity of enzymes, ion channels and vesicular transport, principally through the catalysis of GDP-GTP exchange on heterotrimeric G proteins. They are involved in diverse biological functions including the senses of smell, taste and sight, and the regulation of appetite, digestion, blood pressure, reproduction and inflammation (1) the reason why they are involved in a wide variety of pathologies.
Each cell expresses a few dozen different GPCRs, which implies that its homeostasis can be influenced by numerous transmitters. A particular GPCR is often expressed in several tissues. It can be found in the periphery and in the central nervous system. Its roles in these tissues may be different although the second messengers that result from the initial activation are probably the same. The organ that is possibly most dependent on GPCR activity is the brain, where practically all the GPCRs are expressed. They are involved in synaptic transmission mechanisms and most of our senses depend directly on the activation of specific GPCRs.
GPCRs have proven to be particularly amenable to modulation by small molecule drugs and are the targets of approximately half of the current prescription drugs, as well as the targets of a large number of therapeutics and GPCRs provide opportunities for the development of new drugs with applications in all clinical fields.
CHARACTERISTIC FEATURES OF GPCRS
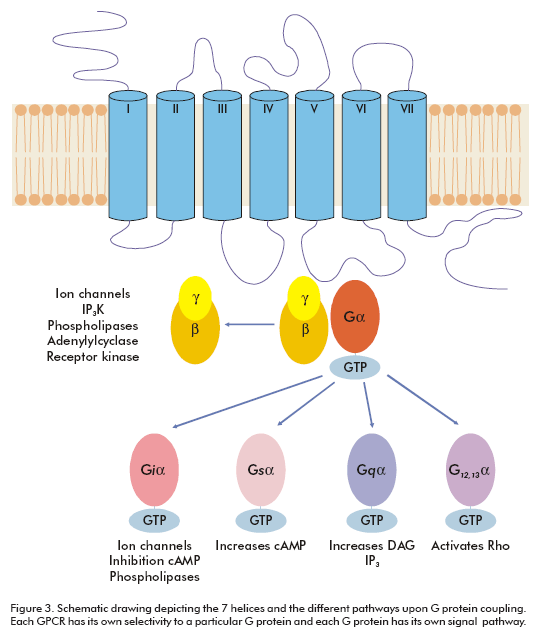
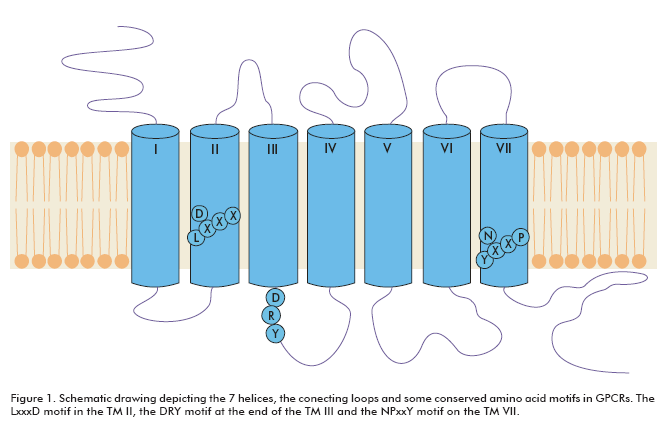
GPCRs are integral membrane proteins with seven transmembrane helices. The N-terminal segment is extracellular and the C-terminal segment is located in the cytosol. The transmembrane (TM) domains are more conserved among GPCRs than the extracellular or intracellular domains. There are several signature amino acid motifs which provide us with their identity as GPCRs; for example, the LxxxD motif in the TM II, the DRY motif at the end of the TM III and the NPxxY motif on the TM VII (Figure 1). Usually, the intracellular domain III (between TM V and TM VI) and the carboxy terminal are considered to play certain roles in G-protein coupling (2).
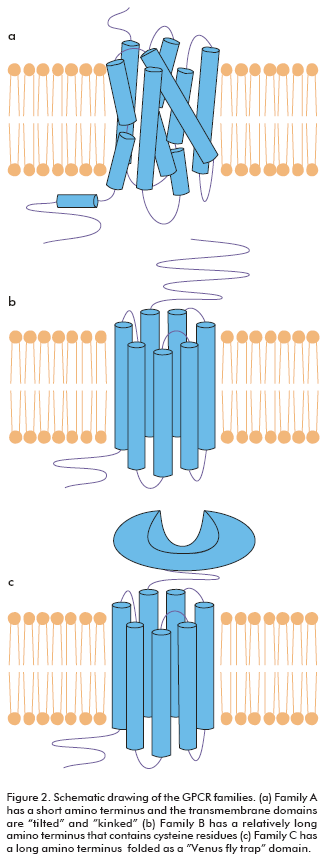
GPCRs are divided into families according to their sequence homology. Family A represents the largest subgroup of receptors and includes catecholamines, neuropeptide, chemokine, glycoproteins, lipid and nucleotide receptors. Family A is characterized by several highly conserved amino acids and a disulphide bridge. Most of these receptors also have a palmitoylated cysteine in the carboxy-terminal tail. Ligand binding within the transmembrane region of the receptor seems to occur mainly in a cavity flanked by TMs III, V, VI and VII. The crystal structure of rhodopsin (3, 4) has indicated that the transmembrane domains of this family are tilted and kinked (Figure 2a). Family B contains receptors for a large number of peptides such as calcitonin, glucagon, gonadotropin-releasing hormone and parathyroid hormone. These receptors are characterized by a relatively long amino terminus that contains six conserved cysteine residues, which presumably form a network of disulphide bridges (Figure 2b). This amino terminus seems to play a key role for most ligands, but it is not sufficient and additional interactions are found in the extracellular loops. Family C is the metabotropic containing the metabotropic glutamate receptors, GABA receptors and the calcium sensor receptor. These receptors are characterized by a long amino terminus and carboxyl tail. The amino terminus is folded as a separate ligand binding domain which is often described as being like a Venus fly trap (Figure 2c) (1, 5, 6).
Ligand binding to GPCRs promote conformational changes leading to G-protein coupling, the initiation of signal transduction pathways and ultimately cellular response. Studies based on electron paramagnetic resonance and fluorescence spectroscopy (7) suggested the need of an outward movement of the cytoplasmic end of TMs III and VI (8, 9), as well as an anti-clockwise rotation of TM VI around its helical axis, when viewed from the extracellular side, for its activation. Other helices probably adjust their positions upon activation as well.
Each GPCR has its own selectivity to G proteins (Figure 3), however, the specific sequences activating each G protein (Gs, Gi, Gq, G12, etc.) are as yet unknown, although there is a proposed theory that basic amino acids are important for G protein coupling (10). Even though it is known that for many classes of receptors constitutive or ligand-induced oligomerization is essential for signaling (11), only a monomeric model for GPCRs is generally accepted. Since the mid- 1990s, many reports have successively shown oligomerization of the GPCRs, examples of this are the H2 histamine receptor (12) and the β2-adrenergic receptor (13, 14). Now, oligomerization is widely accepted as a universal aspect of GPCR biology.
After the first reports of GPCR homo-oligomers, it was shown that some receptor subtypes formed hetero-oligomers, for example AT1-AT2 angiotensin receptors (15) and A1 adenosine-D1 dopamine receptors (16), and that these heteromeric receptors had functional characteristics that differed from homogeneous populations of their constituent receptors. The generation of new properties through hetero-oligomerization indicated a possible mechanism for a generating diversity of functions among GPCRs that had not previously been anticipated (5).
GPCRS IN DRUG DISCOVERY
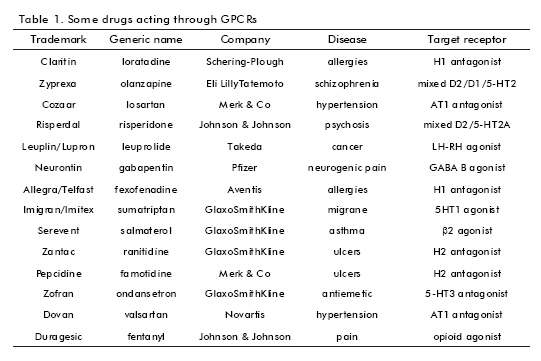
GPCRs have been shown to be excellent targets for pharmaceutical treatments; along with kinases, GPCRs constitute the most widely screened classes of signal transduction targets (17). Many major diseases involve the malfunction of these receptors making them the most important drug target for pharmacological intervention. In particular, the subfamily of biogenic aminebinding GPCRs has provided excellent targets for the treatment of several central nervous system diseases, such as schizophrenia (mixed D2/D1/5-HT2 receptors), psychosis (mixed D2/5-HT2A receptors), depression (5-HT1 receptor), or migraine (5-HT1 receptor). This GPCR subfamily has also provided drug targets for other disease areas such as allergies (H1 receptor), asthma (β2 receptor), ulcers (H2 receptor), or hypertension (α1 antagonist, β1 antagonist) (18) (Table 1).
GPCR agonist or antagonist drugs have been therapeutically successful because of their direct activity on the cell surface (19). GPCRs comprise 50-60% of the drugs now on the market, including about 25% of the 100 top-selling drugs (20). In commercial terms, GPCRs will continue to predominate as drug targets. The total human genome
consists of approximately 35 000 genes. Further analysis suggests that approximately 10% of these genes could be targets for drug intervention in the treatment of diseases. These approximately 3000 genes include those encoding for receptors, ion channels, enzyme inhibitors and GPCRs that might be encoded by 750 of these genes. Almost half of these sequences are likely to encode sensory receptors, leaving around 400 receptors that could be considered as potential drug targets (21). In addition, current GPCR-based drugs only target ~30% of the approximately 200 known GPCRs already identified in the genome, so there are still enormous opportunities for further drug discovery in this field.
Structure-based drug design
Structure-based drug design is widely used in the development of novel drugs. Using structural-based methods it is possible to select compounds with biological activity for synthesis and biological assay. Ligands or target structural information is needed for this approach, which are then divided in ligand- or target based methods.
Ligand-based methods are traditionally used when no protein structure is available. The 2D ligand information can be used to develop new ligands with a structural similarity or the 3D pharmacophore search is used to identify ligands with similar steric and electrostatic features that are recognized at the target binding site and are considered responsible for the biological activity.
Target-based methods require 3D structure of the target and effective scoring procedures. The knowledge of the three-dimensional structure of the protein target is used to identify compounds that can bind to the target with a high specificity, resulting in the inhibition or activation of the target and its effector system.
Ligand-based drug design
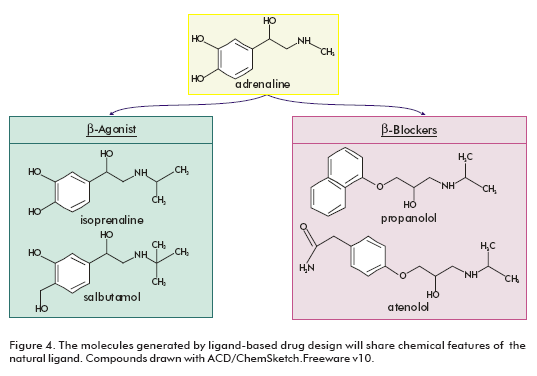
The limited availability of structural data makes ligandbased drug design a very important technique in GPCRs studies. The natural ligand can provide a good starting point in the lead finding process. The aminergic GPCR ligands are extensively used for this purpose (22), because they have the physicochemical requirements for oral absorption (small, moderately lipophilic molecules that tend to exhibit minimal hydrogen bonding potential) (23). Structure-activity relationships (SAR) can be directly derived from natural ligands and their analogues. The resulting pharmacophore models can then be employed for virtual screening to identify lead structures with novel scaffolds. The molecules generated in this manner will share characteristics of the ligand and might have the potential to displace the ligand from the receptor. This is exemplified for the adrenegic β-receptors (Figure 4). Both the ®-agonists (isoprenaline and salbutamol) and the β-blockers (propanolol and atenolol) share significant chemical features with adrenaline (22).
On the other hand, non-aminergic ligands do not exhibit the most desired physicochemical requirements for oral absorption. Especially for peptide-binding GPCRs the identification of a non-peptidic ligand is crucial for drug discovery to avoid the inherent pharmacokinetic problems frequently associated with peptide lead structures like poor oral bioavailability or metabolic instability (22).
However, especially for peptide-binding GPCRs, the screening of diverse or focused compound sets still remains a successful lead finding approach, which has led to the discovery of several potent, non-peptidic GPCRs ligands (24-26). Such compounds have been classified as functional mimetics as they elicit agonist or antagonist activity, but do not necessarily mimic the structure of the native ligand. Examples for the successful use of peptide-derived structure-activity relationship to design non-peptidic GPCR ligands are described for the SST receptor (27), the opiate receptor (28), the thrombin receptor (29), the growth hormone secretagogues receptor (30), and the urotensin II receptor (31).
Ligand-based three-dimensional quantitative structure- activity relationship (or 3D-QSAR) methods and comparative molecular field analyses (CoMFA)(32- 33), have supported the chemical optimization of numerous GPCR lead compounds. Thus, a CoMFA study enables chemical modifications that are beneficial or detrimental for biological activity. There are studies of CoMFA in optimizing GPCR-directed ligands as described, for example, for the dopamine receptors (34-36), the serotonin receptors (37-39), the endothelin receptor (40), and the adenosine receptors (41, 42).
CoMFA models can also be used to recognize molecular features that are responsible for the selectivity of the ligands. A series of aryl piperazines that were active against the 5-HT1A receptor have collateral affinities for the 〈1-adrenergic receptor (43). A separate CoMFA model was derived for each receptor and the comparison of the models indicated that bulky substitutes at the meta position of the aryl moiety would increase selectivity for the 5-HT1A receptor; the a1 receptor, in contrast, does not tolerate large residues at this position. Furthermore, increasing the length of the alkyl chain linking the arylpiperazine with a hydantoin moiety was very beneficial for the desired selectivity.
Privileged structures
The term privileged structure (44) is accepted as a single molecular framework that is able to provide ligands for diverse receptors and it is considered that the modification of such structures could be an alternative in the search for new receptor agonists and antagonists.
This term refers to scaffolds or molecular fragments that seem to reappear in hits, with a relatively high frequency, within a particular group of receptors. The use of privileged structures as a basis for library design has been used to find compounds with good affinity.
Examples of GPCR privileged substructures are biphenyl, 1,1-diphenylmethane, xanthines, 4-arylpiperidines, 4-arylpiperazines, and spiropiperidines (Figure 5). Some of the privileged substructures are not restricted to one GPCR subfamily. Spiropiperidine moiety can be found within ligands of biogenic amine receptors as well as within compounds acting on chemokine and peptide-binding GPCRs. These structures have been notably used for peptide receptors such as somatostatin agonists (Figure 5, structure 1) as well as growth hormone secretagogues (Figure 5, structure 2) (45, 46).
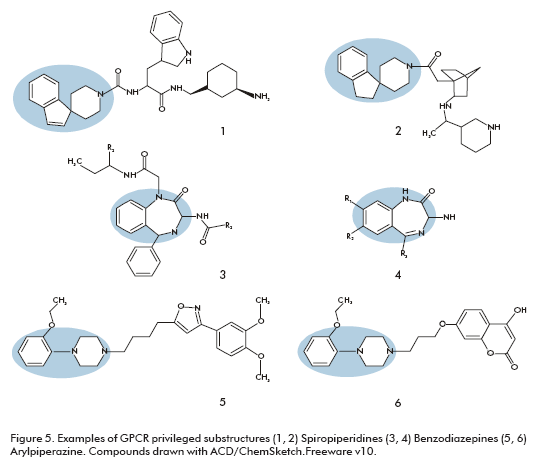
Benzodiazepine based medications are currently in use for several types of central nervous system receptors and in ligands of ion channels and GPCRs throughout the body, and this family is still successfully exploited. Very potent oxytocin antagonists were found by further decorating the two nitrogens of a 1,4-benzodiazepine (Figure 5, structure 3) (47). Similarly, a 1,4-benzodiazepine library (Figure 5, structure 4) was used to identify ligands for cholecystokinin-B from a set of 168 compounds (48).
Arylpiperazines are another very versatile template, in particular for dopaminergic, serotoninergic and adrenergic receptors. A library of 300 discrete arylpiperazines was examined to identify a nanomolar ligand (Figure 5, structure 5), for the D3 receptor showing good selectivity over the corresponding D2 receptor (49). However the use of a promiscuous template can result in hits with binding at other receptors, for example other arylpiperazine molecules (Figure 5, structure 6) showed a good affinity at the D3, 5HT2a and a1Aadrenergic receptors (50). In this case, the lack of selectivity was not a problem because it curiously solved the adverse effects of classical antipsychotics.
Even though the usage of privileged substructures for lead finding offers the chance to quickly identify new lead compounds against novel GPCR targets, receptor selectivity has to be addressed within the lead optimization process, because compounds sharing a privileged substructure often reveal activity against many GPCRs, and could even elicit activities on other yet unknown GPCR mediated mechanism, with adverse side effects.
Target-based drug design
Target-based drug design is another approach for discovering compounds exhibiting biological activity, but it needs the tridimensional structure of the target. In the case of GPCR, that poses a great problem. With the exception of bovine rhodopsin (3, 4), the atomic level structures of other GPCRs, in particular for potential GPCRs drugs, are yet unknown. Rhodopsin is unique among GPCRs; it consists of two building blocks, an opsin protein and a reversibly covalently bound ligand, retinal (Figure 6). The other available structure of a 7TM (7 transmembrane helices) protein is that of bacteriorhodopsin (51), but bacteriorhodopsin is not a GPCR and in contrast to the beliefs of many authors for a large number of years, its 3D structure is significantly different from that of rhodopsin (6).
In the absence of experimentally determined structures for GPCRs, computational protein modeling becomes an important approach for structure-based drug discovery for GPCRs. 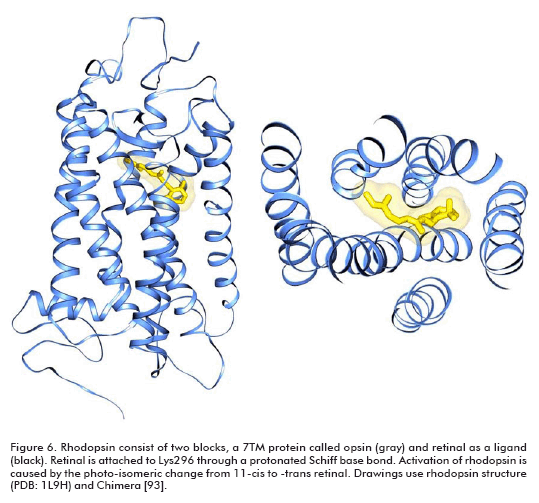
Modeling GPCRs
The most straightforward approach to determine the 3D structure of GPCRs is homology modeling. Although bovine rhodopsin reveals a low sequence similarity to other GPCRs, the specific arrangement of the 7TM helices stabilized by a series of intramolecular interactions mediated by several backbone and side-chain atoms seems to be conserved among the Family A receptors. Rhodopsin thus represents an improved structural template for the understanding of experimental data available for related 7TM receptors and for generating improved molecular models of other Family A receptors (6).
Homology modeling methods for GPCRs with sufficient accuracy for structure-based drug design would have an enormous impact on drug discovery. This goal faces several serious challenges. Many targets of interest for drug development share rather low sequence identity (30%) with rhodopsin. The transmembrane helices can frequently be aligned with reasonable certainty aided by certain highly conserved residues (e.g., DRY on TMIII, NPxxY on TMVI) and GPCR models based on a template with an identity of 20-30% can thus be expected to be of greater accuracy than when modeling other types of proteins based on a template with a low-sequence identity.
Rhodopsin-based homology models have been developed for the dopamine D2 receptor (52), opioid receptors (53) and the α2 adrenergic receptor (54).
Not only homology modeling can be used to obtain GPCRs models. The PREDICT algorithm (55, 56) is a de novo approach for modeling the 3D structure of any GPCR that is not based on a homology to a known structure of rhodopsin or bacteriorhodopsin. The PREDICT method relies on the primary sequence of the receptor itself and on structural constraints imposed by the membrane environment. PREDICT is based on the physicochemical properties of a single sequence and is therefore substantially different from the existing modeling approaches that rely on known structures or multiple sequence alignment. It was demonstrated that PREDICT was able to reproduce the known experimental structure of rhodopsin (6). The quality of the PREDICT models for drug discovery purposes was validated by their successful utilization in virtual screening (57). Examples of this are Dopamine D2, neurokinin NK1, and neuropeptide Y Y1 receptors (58).
Threading assembly refinement (TASSER) is another method that was recently developed (59). This methodology combines threading and ab-initio algorithms to span the homologous to non-homologous regimens. Only the sequence of the given GPCR is needed and no other extrinsic knowledge (e.g., active sites and binding regions, experimental restraints, etc.) is incorporated into the structure prediction approach. Also, distinct from many other GPCR modeling methods that only attempt to model the TM helical regions (58, 60, 61), TASSER generates reasonable predictions for the loop regions. This is especially important in GPCR modeling as the extracellular loops are often critical in determining ligand specificity (62-64). Therefore, full-length TASSER models might offer substantial advantages over traditional comparative modeling methods and are likely to be of greater aid in understanding the ligand and signaling interactions of GPCRs.
On our experience the modeling of a GPCR is not, and will never be, an easy task as long as no new crystal structures are elucidated, having only one template to choose from and this being from a single family, all the prediction methods can only be validated using the structure of rhodopsin and this could be strongly biased when used for the prediction of GPCRs from the rest of the families.
Docking and Virtual screening with GPCRs models
A three-dimensional model of the human melanocortin 4 receptor (hMC4R) was constructed (65), using the transmembrane helices and the C terminal domain of bovine rhodopsin, and simulating both intracellular and extracellular loop domains on homologous loop regions in other proteins of known 3D structure and further refining the structure by minimization and dynamic calculations. The model was tested by docking with a triplet peptide (RFF) ligand. The ligand-receptor interactions found, were consistent with mutational and biochemical data.
Another study was done (66) using only the helical centers of the rhodopsin structure for predicting the 3D structures of rhodopsin and of the b1 adrenergic receptor. The binding mode of adrenaline docked into this b1 adrenergic receptor model was in good agreement with the experimental data.
In another approach (67), it was suggested that GPCRs models based on the rhodopsin X-ray structure are therefore expected to be closer to their inactive form than to their activated, agonist-bound state. Therefore, they have optimized rhodopsin-based models for the agonist and antagonist by energy minimization with one known agonist or antagonist docked to the active site, respectively, even though in the case of an agonist it might not be enough to generate correct models of an activated state of a GPCR. The ligandbound models of several receptors generated this way, including dopamine D3, vasopressin V1a, β2 adrenergic and opioid receptors, proved successful in computational screening tests. They also did cross-docking experiments and showing that virtual screening against the new agonist-bound states of related GPCRs is selective enough to distinguish not only true ligands from randomly chosen drug-like molecules but also true hits from chemically related inactive compounds (67). Applied to three human GPCRs, such receptor models are accurate enough for discriminating known agonists from randomly chosen drug-like molecules. Most importantly, they were able to retrieve in the virtual hit lists true agonists whose chemical structures had not previously been used for generating the pharmacophore and reûning the receptor model (67).
Virtual screening based on GPCR models may be particularly important in cases when either limited or no ligand information is available (18). This is true for most of the pharmaceutically relevant GPCRs, for which only the endogenous ligand is known. Recent publications report successful applications of GPCR models in virtual screening (67-70), indicating the general relevance of GPCR models and their usefulness for structure-based drug design. For example, a virtual screening for the D3 receptor using a homology model of this receptor was done and eight, out of 20 experimentally tested compounds showed Ki values better than 1 µM (70).
Different virtual screening strategies were compared for the identification of biogenic amine-binding GPCR antagonists, starting from virtual libraries, consisting of the antagonist of these target receptors (the a1A, 5HT2A, D2, and M1 receptors) and additional drug-like molecules (18). And the results were also in agreement with the expectations.
In another attempt to explore the suitability of GPCR homology models for the purpose of virtual screening, a homology model for the a1A receptor was generated (69). Applying two-dimensional (2D) queries and a 3D-pharmacophore model as a prefilter, they docked 23 000 ligands into the a1A receptor homology model, 37 out of the 80 compounds that were selected for experimental testing, showed a Ki value better than 10 μM, and 24 of these compounds were even binding in the sub-micromolar range. The hit rates achieved with these models were similar to those typically reached when the target protein is given by a crystal structure, suggesting that docking into rhodopsin-based GPCR models, is indeed a feasible approach for the identification of novel ligands.
Even when it is widely accepted that docking to models is more challenging and less successful than docking to crystallographic structures, surprisingly little work has been done to quantify the accuracy of docking to homology models or to improve the existing methods particularly for docking to homology models. The true fact is that any reasonable structure could produce a good hit rate on ligand design or screening if one is careful enough in the selection of the docking experimental parameters.
ORPHAN GPCRS
Even though GPCRs have been intensely investigated as potential drug targets, their structural and functional diversity (71, 72), still offer opportunities to develop novel drugs. The analyses of the human genomic sequence suggest that there may be 750 human GPCR-encoding genes, of which approximately 160 cannot be functionally characterized either on the basis of sequence homology or by association with known endogenous ligands (21). These are referred to as orphan GPCRs (oGPCRs) which bind (as yet) unknown ligands (73, 74).
Reverse pharmacology is an approach that can be used for GPCRs deorphanization. Based on the idea that GPCRs are targets of neurotransmitters, peptides, hormones and other transmitters, it can be expected that orphan GPCRs are also activated by transmitter molecules. Then the orphan GPCR is used as a target to test potential transmitters (73, 75).
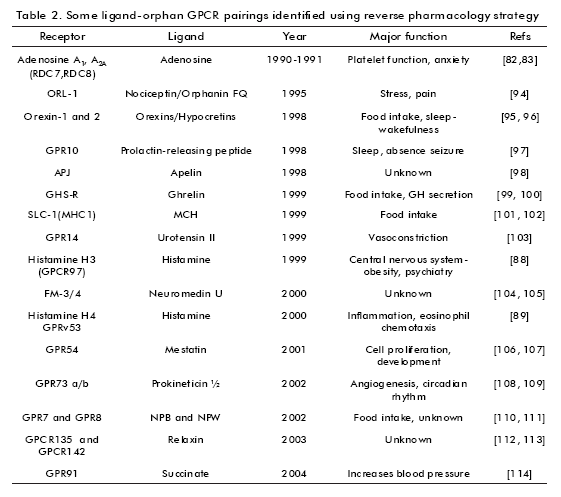
The first efforts to identify ligands for orphan GPCRs began in the mid-1980s. At that time, the number of known potential transmitters was large. This became reverse pharmacology in an important approach to this aim. The first successful deorphanization of orphan GPCRs were reported in 1988 (5- HT1A receptors (76) and dopamine D2 receptors (77)). The strategies used were the same, that is, the orphan GPCR was expressed by DNA transfection in eukaryotic cells, membranes of these cells were then used as targets to determine the binding of potential transmitters. During the first part of the 1990s, the application of the reverse pharmacology strategy led to the pharmacological characterization of many GPCRs (Table 2).
Reverse pharmacology has been adapted to allow for the screening of a large battery of potential transmitters on batteries of orphan GPCRs by using high-throughput screening techniques. This has made it possible to match several dozens of orphan GPCRs to their ligands (19, 73). But all these ligands had been previously discovered and there was a need to identify new transmitters.
While over the years, numerous orphan GPCRs have been matched to specific ligands, there are over one hundred GPCRs that do not bind any known transmitters (78). In the mid-1990s a parallel approach was devised to use orphan GPCRs as targets to find novel, still non-described transmitters. This has been termed the orphan receptor strategy (73, 79). The method consists of expressing an orphan GPCR by transfection into eukaryotic cell lines, preparing a tissue extract expected to contain the transmitter specific to the orphan receptor and monitoring the activation of the GPCR by applying finely fractioned tissue extract over these engineered cell lines. The activation of the orphan GPCR is monitored by measuring second messenger responses. Positive extracts are fractionated biochemically until the active component is isolated and characterized (78). This approach has led to the discovery of dozens of bioactive peptides. The orphan receptor strategy was first applied in 1995 to the discovery of a novel neuropeptide called orphanin FQ or nociceptin (or OFQ/N).
Traditionally the existence of a transmitter was postulated on the basis of a particular physiological response and was isolated using that response as an assay. The orphan receptor strategy reverses this approach and allows the isolation of transmitters with unknown physiology and linkage to a disease process. The success of this approach, however, is a big leap towards understanding the transmitter system by using the receptor as a vehicle to unravel its physiological function (78).
Nowadays finding the natural ligand of an orphan GPCR, is equal to that of finding a novel transmitter. However, finding the natural ligands of an orphan GPCR is a challenge, since neither the biochemical properties of the ligand nor the response that receptor activation will induce are known.
The G-protein signaling pathway is often unknown, and to maximize the chance of success the assay system must be as generic as possible to allow for the detection of a wide range of signaling mechanisms, but also to be amenable to high throughput screening so that the activity of a large number of ligands can be readily measured. Such assay systems rely mainly on measuring changes in intracellular cAMP or calcium levels, either directly or through the use of reporter gene assays (21) and the presence of endogenous receptors, which result in background responses to ligands, can be avoided by engineered cell strains that have had the endogenous GPCR genetically deleted (80, 81).
Other approaches can be used in GPCR deorphanization, e.g. determining the relationship between receptor- expression patterns and the expression pattern of a putative ligand, thus matching a candidate gene with a pharmacologically identified receptor. This led to the identification of orphan receptors RDC7 and RDC8 as adenosine A1 and A2A receptors (82, 83).
Sometimes sequence homology can be helpful in GPCR deorphanization. The DNA sequence of the orphan receptor is compared with the sequences of liganded receptors, and where they are closely related it is sometimes possible to predict the likely cognate ligand of the orphan receptor. This approach has been successful in some cases. The initial demonstration that OGR-1 could act as a high affinity receptor for the lipid sphingosyl phosphorylcholine (84) probably facilitated the more recent identification of two related orphan GPCRs, TDAG-8 and G2A, as receptors for the lipids psychosine (85) and lysophosphatidyl choline (86), respectively. The ligands for the fourth member of this receptor subfamily, GPR 4, have recently been identified. As would be predicted from sequence homology, the ligands for GPR4 are lysophosphatidyl choline and sphingosyl phosphorylcholine (87). However, using amino acid sequence identity as the basis for such experiments can be misleading as recently demonstrated following the cloning of histamine H3 (88) and H4 (89) receptors. Both these receptors have the lowest recorded identity to other members of their receptor family (~20% overall to H1 and H2), which highlights the fact that it is not always possible to make accurate predictions. Another example is a receptor originally known as P2Y7 (BLT1) that was thought to be a nucleotide receptor based on its similarity to P2Y receptors, but it was shown to be activated by an unrelated ligand, leukotriene B4 (90). Sequence homology gives only an indication as to the nature of the likely ligand, but it is not yet possible to accurately predict which ligand is likely to bind to a novel receptor simply from an analysis of the sequence of that receptor.
Homology modeling and ligand docking might even be helpful for the deorphanization of GPCRs. Docking into GPCR homology models can be a useful approach for lead finding by virtual screening when either little or no information on the active ligands is available (18). Once the generation of reliable GPCR structure models of the activated receptor state becomes possible (91), molecular docking might even provide an opportunity for the identification of novel agonists.
Some work has been done to virtually screen for ligands of orphan GPCRs (92). This approach may be used to contribute to the functional characterization of orphan GPCRs by identifying potential cognate ligands, thereby providing clues to guide the therapeutic regulation of important signaling pathways in the cell. The advantage of this approach is the simplicity of the required input data: proteins are described using only physicochemical properties of primary amino acid sequences, and ligand features are based on the two-dimensional connectivity between constituent atoms and atomic properties. In its application, large numbers of chemical compounds may be screened against a particular orphan GPCR sequence, with a ranked list of putative high-affinity ligands generated automatically on output.
The deorphanization of GPCRs has revolutionized the discovery of novel transmitters and in turn these have revolutionized many fields of biomedical research in which they have been involved. For example, the novel neuropeptides found as ligands of orphan GPCRs have changed our understanding of the mechanisms that regulate sleep or food intake. Many of the deorphanized GPCRs are targets of drug development programs.
NEW GPCR STRUCTURES
From October 2007 to October 2008, during the editing process of this review, new GPCR structures were released to the public by the RCSB, and they were considered a major breakthrough for the Structural Biology. Three proteins from family A: the human β2 adrenergic receptor (115-117), the human A2A adenosine receptor (118) and the β1 adrenergic receptor (119) from the common turkey were engineered and crystallized using novel methods yielding high resolution structures (2rh1, 2r4r, 2r4s, 3d4s, 3eml, 2vt4).
Although there are not yet models reported to being built using any of them as a template, the solely existence of the structures is a very important factor to improve the knowledge about the structural regularities and differences amongst the GPCRs, and indeed will help us to build better models.
We certainly hope that in the near future we could expect more structures from other GPCR families.
CONCLUSIONS
GPCRs are regarded as the most important molecules in the field of drug discovery and design, their role as receptors in many of the basic processes on the organism and their presence on the surface of cells on all tissues make them excellent targets. Much effort is needed, however, in the deorphanization of GPCRs, matching all currently known molecules with a ligand. There are several initiatives in this field, but their difficulty makes the progress very slow. Despite of the fact that the structure of a single family is known, research has progressed, using combined structure-based techniques. There are several groups attempting to purify, fold and crystallize GPCRs with important breakthroughs, and structures could be expected in the near future. This would undoubtedly change the course of structure-based design for GPCR targets.
REFERENCES
1. Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev 2000;21:90-113.
2. Oliveira LPA, Vriend G. A common motif in G-protein-coupled seven transmembrane helix receptors. J Comput Aided Mol Des 1993;7:649-58.
3. Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000;289:739-45.
4. Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol 2004;343:1409-38.
5. George SR, ODowd BF, Lee SP. Gprotein- coupled receptor oligomerization and its potencial for drug discovery. Nat Rev Drug Discov 2002;1:808-20.
6. Klabunde T, Hessler G. Drug Design Strategies for Targeting G-Protein-Coupled Receptors. Chem BioChem 2002;3:928-44.
7. Farrens DL, Altenbach C, Yang K, Hubell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 1996;274:768-70.
8. Dunham TD, Farrens DL. Conformational changes in Rhodopsin. J Biol Chem 1999;274:1683-90.
9. Jensen AD, Guarnieri F, Rasmussen SGF, Asmar F, BallesterosJA, Gether U. Agonist-induced conformational changes at the cytoplasmatic side of transmembrane segment 6 in the B2- adrenergic receptor mapped by site-selective fluorescent labeling. J Biol Chem 2001;276:9279-90.
10. Okamoto T, Murayama Y, Hayashi Y, Inagaki M, Ogata E, Nishimoto I. Identification of a Gs activator region of the beta 2-adrenergic receptor that is autoregulated via protein kinase A-dependent phosphorylation. Cell 1991;67:723-30.
11. Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell 1995;80:213-23.
12. Fukushima Y, Asano T, Saitoh T, Anai M, Funaki M, Ogihara T, et al. Oligomer formation of histamine H2 receptors expressed in Sf9 and COS7 cells. FEBS Lett 1997;409:283-6.
13. Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, Bouvier M. A peptide derived from a a2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem 1996;271:16384-92.
14. Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, Bouvier M. Detection of a2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc Natl Acad Sci USA 2000;97:3684-9.
15. AbdAlla S, Lother H, Abdel-Tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem 2001;276:39721-6.
16. Gines S, Hillion J, Torvinen M, Le Crom S, Casado V, Canela EI, et al. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci USA 2000;97:8606-11.
17. Auld DS, Diller D, Ho KK. Targeting signal transduction with large combinatorial collections. Drug Discov Today 2002;7:1206-13.
18. Evers A, Hessler G, Matter H, Klabunde T. Virtual Screening of Biogenic Amine- Binding G-Protein Coupled Receptors: Comparative Evaluation of Protein- and Ligand-Based Virtual Screening Protocols. J Med Chem 2005;48:5448-65.
19. Howard A, McAllister G, Feighner S, Liu Q, Nargund R, Van de Ploeg L, Patchett A. Orphan G protein-coupled receptors and natural ligand discovery. Trends Pharmacol Sci 2001;22:132-40.
20. Drews J. Drug discovery: A historical perspective. Science 2000;287:1960-4.
21. Wise A, Gearing K, Rees S. Target validation of G-protein coupled receptors. Drug Discov Today 2002;7:235-46.
22. Beaumont K, Schmid E, Smith DA. Oral delivery of G protein-coupled receptor modulators: An explanation for observed class diference. Bioorg Med Chem Lett 2005;15:3658-64.
23. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46:3-26.
24. Freidinger RM. Nonpeptidic ligands for peptide and protein receptors. Curr Opin Chem Biol 1999;3:395-406.
25. Beeley NRA. Can peptides be mimicked? Drug Discov Today 2000;5:354-63.
26. Gurrath M. Peptide-binding G protein-coupled receptors: New opportunities for drug design. Curr Med Chem 2001;8:1605-48.
27. Veber DF. Design and discovery in the development of peptide analogues. In: Smith JA, Rivier JE, editors. Peptides, Chemistry, and Biology. Proceedings of the 12th American Peptide Symposium;1992; ESCOM, Leiden, Netherlands.
28. Alfaro-López J, Okayama T, Hosohata K, Davis P, Porreca F, Yamamura HI, Hruby VJ. Exploring the structure-activity relationships of [1-(4-tert-butyl-3'-hydroxy) benzhydryl-4-benzylpiperazine] (SL- 3111), a high-affinity and selective deltaopioid receptor nonpeptide agonist ligand. J Med Chem 1999;42:5359-68.
29. Alexopoulos K, Panagiotopoulos D, Mavromoustakos T, Fatseas P, Paredes-Carbajal MC, Mascher D, et al. Design, synthesis, and modeling of novel cyclic thrombin receptor-derived peptide analogues of the Ser42-Phe-Leu-Leu-Arg46 motif sequence with fixed conformations of pharmacophoric groups: importance of a Phe/Arg/NH2 cluster for receptor activation and implications in the design of nonpeptide thrombin receptor mimetics. J Med Chem 2001;44:328-39.
30. Huang P, Loew GH, Funamizu H, Mimura M, Ishiyama N, Hayashida M, et al. Rational design, discovery, and synthesis of a novel series of potent growth hormone secretagogues. J Med Chem 2001;44:4082-91.
31. Flohr S, Kurz M, Kostenis E, Brkovich A, Fournier A, Klabunde T. Identification of nonpeptidic urotensin II receptor antagonists by virtual screening based on a pharmacophore model derived from structure-activity relationships and nuclear magnetic resonance studies on urotensin II. J Med Chem 2002;45:1799- 805.
32. Cramer RD, Patterson DE, Bunce JD. Comparative Molecular Field Analysis (CoMFA). 1. Effect of Shape on Binding of Steroids to Carrier Proteins. J Am Chem Soc 1988;110:5959-67.
33. Kim KH, Greco G, Novellino EA. Critical Review of Recent CoMFA Applications. Perspect Drug Discov Des 1998;12-14:257-315.
34. Lanig H, Utz W, Gmeiner P. Comparative molecular field analysis of dopamine D4 receptor antagonists including 3-[4-(4-chlorophenyl)piperazin-1-ylmethyl] pyrazolo[1,5-a]pyridine (FAUC 113), 3-[4-(4-chlorophenyl)piperazin-1- ylmethyl]-1H-pyrrolo-[2,3-b]pyridine (L- 745,870), and clozapine. J Med Chem 2001;44:1151-7.
35. Hoffman B, Cho SJ, Zheng W, Wyrick S, Nichols DE, Mailman RB, Tropsha A. Quantitative structure-activity relationship modeling of dopamine D(1) antagonists using comparative molecular field analysis, genetic algorithms-partial least squares, and K nearest neighbor methods. J Med Chem 1999;42:3217-26.
36. McGaughey GB, Mewshaw RE. Application of comparative molecular field analysis to dopamine D2 partial agonists. Bioorg Med Chem 1999;7:2453-6.
37. Gaillard P, Carrupt PA, Testa B, Schambel P. Binding of arylpiperazines, (aryloxy) propanolamines, and tetrahydropyridylindoles to the 5-HT1A receptor: contribution of the molecular lipophilicity potential to three-dimensional quantitative structureaffinity relationship models. J Med Chem 1996;39:126-34.
38. López-Rodríguez ML, Benhamu B, Viso A, Morcillo MJ, Murcia M, Orensanz L, Alfaro MJ, Martin MI. Benzimidazole derivatives. Part 1: Synthesis and structure-activity relationships of new benzimidazole- 4-carboxamides and carboxylates as potent and selective 5-HT4 receptor antagonists. Bioorg Med Chem 1999;7:2271-81.
39. López-Rodríguez ML, Murcia M, Benhamu B, Viso A, Campillo M, Pardo L. 3-D-QSAR/CoMFA and recognition models of benzimidazole derivatives at the 5- HT(4) receptor. Bioorg Med Chem Lett 2001;11:2807-11.
40. Chen Q, Wu CD, Maxwell D, Krudy GA, Dixon RAF, You TJ. A 3D QSAR analysis of in vitro binding affinity and selectivity of 3-isoxazolyl-sulfonyl-aminothiophenes as endothelin receptor antagonists. Quant Struct-Act Relat 1999;18:124-33.
41. Siddiqi SM, Pearlstein RA, Sanders LH, Jacobson KA. Comparative molecular field analysis of selective A3 adenosine receptor agonists. Bioorg Med Chem 1995;3:1331-43.
42. Rieger JM, Brown ML, Sullivan GW, Linden J, Macdonald TL. Design, synthesis, and evaluation of novel adenosine A2A receptor agonists. J Med Chem 2001;44:531-9.
43. López-Rodríguez ML, Rosado ML, Benhamu B, Morcillo MJ, Fernández E, Schaper KJ. Synthesis and structure-activity relationships of a new model of arylpiperazines. 2. Three-dimensional quantitative structure-activity relationships of hydantoin-phenylpiperazine derivatives with affinity for 5-HT1A and alpha 1 receptors. A comparison of CoMFA models. J Med Chem 1997;40:1648-56.
44. Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J Med Chem 1988;31:2235-46.
45. Patchett AA, Nargund RP. Privileged structures -an update. Annu Rep Med Chem 2000;35:289-98.
46. Palucki BL, Feighner SD, Pong S-S, McKee KK, Hreniuk DL, Tan C, et al. Spiro(indoline-3,40 -piperidine) growth hormone secretagogues as ghrelin mimetics. Bioorg Med Chem Lett 2001;11:1955-7.
47. Evans B, Pipe A, Clark L, Banks M. Identification of a potent and selective oxytocin antagonist, from screening a fully encoded differential release combinatorial chemical library. Bioorg Med Chem Lett 2001;11:1297-300.
48. Lattman E, Billington DC, Poyner DR, Arayarat P, Howitt SB, Lawrence S, Offel M. Combinatorial solid phase synthesis of multiply substituted 1,4-benzodiazepines and affinity studies on the CCK2 receptor. Drug Des Discov 2002;18:9-21.
49. Cha MY, Choi BC, Kang KH, Pae AN, Choi KI, Cho YS, et al. Design and synthesis of a piperazinyl alkylisoxazole library for subtype selective dopamine receptor ligands. Bioorg Med Chem Lett 2002;12:1327-30.
50. González-Gómez JC, Santana L, Uriarte E, Brea J, Villazon M, Loza MI, et al. New aryl-piperazine derivatives with high afinity for a1A, D2, and 5-HT2A receptors. Bioorg Med Chem Lett 2003;13:175-8.
51. Pebay-Peyroula E, Rummel G, Rosenbusch JP, Landau EM. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 1997;277:1676-81.
52. Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol 2001;60:1-19.
53. Strahs D, Weinstein H. Comparative modeling and molecular dynamics studies of the delta, kappa and mu opioid receptors. Protein Eng 1997;10:1019-38.
54. Green SA, Spasoff AP, Coleman RA, Johnson M, Liggett SB. Sustained activation of a G protein- coupled receptor via anchored agonist binding. Molecular localization of the salmeterol exosite within the 2-adrenergic receptor. J Biol Chem 1996;271:24029-35.
55. Becker OM, Shacham S, Marantz Y, Noiman S. Modeling the 3Dstructure of GPCRs: advances and application to drug discovery. Curr Opin Drug Discov Dev 2003;6:353-61.
56. Shacham S, Topf M, Avisar N, Glaser F, Marantz Y, Bar-Haim S, et al. Modeling the 3D structure of GPCRs from sequence. Med Res Rev 2001;21:472-83.
57. Shoichet BK, McGovern SL, Wei B, Irwin JJ. Lead discovery using molecular docking. Curr Opin Chem Biol 2002;6:439-46.
58. Shacham S, Marantz Y, Bar-Haim S, Kalid O, Warshaviak D, Avisar N, et al. PREDICT modeling and In-Silico screening for G-Protein coupled receptors. Proteins 2004;57:51-86.
59. Zhang Y, DeVries ME, Skolnick J. Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Comput Biol 2006;2:88-99.
60. Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, Bar-Haim S, Warshaviak D, Fichman M, Noiman S. G protein-coupled receptors: in silico drug discovery in 3D. Proc Natl Acad Sci USA 2004;101:11304-9.
61. Sale K, Faulon JL, Gray GA, Schoeniger JS, Young MM. Optimal bundling of transmembrane helices using sparse distance constraints. Prot Sci 2004;13:2613-27.
62. Shi L, Javitch JA. The binding site of aminergic G protein-coupled receptors: The transmembrane segments and second extracellular loop. Annu Rev Pharmacol Toxicol 2002;42:437-67.
63. Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, et al. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 2000;407:971-7.
64. Du P, Salon JA, Tamm JA, Hou C, Cui W, Walker MW, et al. Modeling the G-protein coupled neuropeptide Y Y1 receptor agonist and antagonist binding sites. Prot Eng 1997;10:109-17.
65. Yang X, Wang Z, Dong W, Ling L, Yang H, Chen R. Modeling and docking of the threedimensional structure of the human melanocortin 4 receptor. J Protein Chem 2003;22:335-44.
66. Vaidehi N, Floriano WB, Trabanino R, Hall SE, Freddolino P, Choi EJ, et al. III. Prediction of structure and function of G protein-coupled receptors. Proc Natl Acad Sci USA 2002;99:12622-7.
67. Bissantz C, Bernard P, Hibert M, Rognan D. Protein-based virtual screening of chemical databases. II. Are homology models of Gprotein coupled receptors suitable targets? Proteins 2003;50:5-25.
68. Evers A, Klebe G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J Med Chem 2004;47:5381-92.
69. Evers A, Klabunde T. Structure-based drug discovery using GPCR homology modeling: successful virtual screening for antagonists of the alpha1A adrenergic receptor. J Med Chem 2005;48:1088-97.
70. Varady J, Wu X, Fang X, Min J, Hu Z, Levant B, et al. Molecular modeling of the three-dimensional structure of dopamine 3 (D3) subtype receptor: discovery of novel and potent D3 ligands through a hybrid pharmacophore- and structure-based database searching approach. J Med Chem 2003;46:4377-92.
71. Hamm HE. The Many Faces of G Protein Signaling. J Biol Chem 1998;273:669-72.
72. Ji TH, Grossmann M, Ji I. G Protein- coupled Receptors. I. Diversity of receptor-ligand interactions. J Biol Chem 1998;273:17299-302.
73. Civelli O, Nothacker HP, Saito Y, Wang Z, Lin SH, Reinscheid RK. Novel neurotransmitters as natural ligands of orphan G-proteincoupled receptors. Trends Neurosci 2001;24:230-37.
74. Im D-S. Orphan G Protein-Coupled Receptors and Beyond. Jpn J Pharmacol 2002;90:101-6.
75. Libert F, Vassart G, Parmentier M. Current developments in G-protein-coupled receptors. Curr Opin Cell Biol 1991;3:218-23.
76. Fargin A, Raymond Jr, Lohse MJ, Kobilka BK, Caron MG, Lefkowitz R. The genomic clone G-21 which resembles a beta-adrenergic receptor sequence encodes the 5-HT1A receptor. Nature 1988;335:358-60.
77. Bunzow Jr, Van Tol HH, Grandy DK, Albert P, Salon J, Christie M, et al. Cloning and expression of a rat D2 dopamine receptor cDNA. Nature 1988;336:783-7.
78. Lin SH, Civelli O. Orphan G protein-coupled receptors: targets for new therapeutic interventions. Ann Med 2004;36:204-4.
79. Civelli O. Functional genomics: the search for novel neurotransmitters and neuropeptides. FEBS Lett 1998;430:55-8.
80. Broach JR, Thorner J. High-throughput screening for drug discovery. Nature 1996;384:14-6.
81. Brown AJ, Dyos SL, Whiteway MS, White JH, Watson MA, Marzioch M, et al. Functional coupling of mammalianreceptors to the yeast mating pathway using novel yeast/mammalian G protein alpha-subunit chimeras. Yeast 2000;16:11-22.
82. Maenhaut C, Van Sande J, Libert F, Abramowicz M, Parmentier M, Vanderhaegen JJ, et al. RDC8 codes for an adenosine A2 receptor with physiological constitutive activity. Biochem Biophys Res Commun 1990;173:1169-78.
83. Libert F, Schiffmann SN, Lefort A, Parmentier M, Gerard C, Dumont JE, et al. The orphan receptor cDNA RDC7 encodes an A1 adenosine receptor. EMBO J 1991;10:1677-82.
84. Xu Y, Zhu K, Hong G, Wu W, Baudhuin LM, Xiao Y, Damron DS. Sphingosylphosphorylcholine is a ligand for ovarian cancer G protein-coupled receptor 1. Nat Cell Biol 2000;2:261-7.
85. Im DS, Heise CE, Nguyen T, ODowd BF, Lynch KR. Identification of a molecular target of psychosine and its role in globoid cell formation. J Cell Biol 2001;153:429-34.
86. Kabarowski JHS, Zhu K, Le LQ, Witte ON, Xu Y. Lysophosphatidylcholine as a ligand for the immunoregulatory receptor G2A. Science 2001;293:702-5.
87. Zhu K, Baudhuin LM, Hong G, Williams FS, Cristina KL, Kabarowski JHS, et al. Sphingosylphosphorylcholine and lysophosphatidylcholine are ligands for the G protein-coupled receptor GPR4. J Biol Chem 2001;276:41325-35.
88. Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, et al. Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol 1999;55:1101-7.
89. Oda T, Morikawa N, Saito Y, Masuho Y, Matsumoto S. Molecular characterization of a novel type of histamine receptor preferentially expressed in leukocytes. J Biol Chem 2000;275:36781-6.
90. Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature 1997;387:620-4.
91. Gouldson PR, Kidley NJ, Bywater RP, Psaroudakis G, Brooks HD, Diaz C, et al. Toward the active conformations of rhodopsin and the beta 2-adrenergic receptor. Proteins 2004;56:67-84.
92. Bock Jr, Gough DA. Virtual Screen for Ligands of Orphan G Protein-Coupled Receptors. J Chem Inf Model 2005;45:1402-14.
93. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605-12.
94. Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 1 95; 377:532-5.
95. Sutcliffe JG, de Lecea L. The hypocretins: setting the arousal threshold. Nat Rev Neurosci 2002;3:339-49.
96. Smart D, Haynes AC, Williams G, Arch Jr. Orexins and the treatment of obesity. Eur J Pharmacol 2002;440:199-212.
97. Lin SH, Arai AC, Espana RA, Berridge CW, Leslie FM, Huguenard Jr, et al. Prolactin-releasing peptide (PrRP) promotes awakening and suppresses absence seizures. Neuroscience 2002;114:229-38.
98. Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun 1998;251:471-6.
99. Hosoda H, Kojima M, Kangawa K. Ghrelin and the regulation of food intake and energy balance. Mol Interv 2002;2:494-503.
100. Muccioli G, Tschop M, Papotti M, Deghenghi R, Heiman M, Ghigo E. Neuroendocrine and peripheral activities of ghrelin: implications in metabolism and obesity. Eur J Pharmacol 2002;440:235-54.
101. Saito Y, Nothacker HP, Wang Z, Lin SH, Leslie F, Civelli O. Molecular characterization of the melanin-concentrating-hormone receptor. Nature 1999;400:265-9.
102. Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melaninconcentrating hormone are hypophagic and lean. Nature 1998;396:670-4.
103. Ames RS, Sarau HM, Chambers JK, Willette RN, Aiyar NV, Romanic AM, et al. Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature 1999;401:282-6.
104. Szekeres PG, Muir AI, Spinage LD, Miller JE, Butler SI, Smith A, et al. Neuromedin U is a potent agonist at the orphan G proteincoupled receptor FM3. J Biol Chem 2 00; 275:20247-50.
105. Kojima M, Haruno R, Nakazato M, Date Y, Murakami N, Hanada R, et al. Purification and identification of neuromedin U as an endogenous ligand for an orphan receptor GPR66 (FM3). Biochem Biophys Res Commun 2000;276:435-8.
106. Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature 2001;411:613-7.
107. Funes S, Hedrick JA, Vassileva G, Markowitz L, Abbondanzo S, Golovko A, et al. The KiSS-1 receptor GPR54 is essential for the development of themurine reproductive system. Biochem Biophys Res Commun 2003;312:1357-63.
108. LeCouter J, Lin R, Ferrara N. Endocrine gland-derived VEGF and the emerging hypothesis of organ-specific regulation of angiogenesis. Nat Med 2002;8:913-7.
109. Cheng MY, Bullock CM, Li C, Lee AG, Bermak JC, Belluzzi J, et al. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature 2002;417:405-10.
110. Shimomura Y, Harada M, Goto M, Sugo T, Matsumoto Y, Abe M, et al. Identification of neuropeptide W as the endogenous ligand for orphan G-protein- coupled receptors GPR7 and GPR8. J Biol Chem 2002;277:35826-32.
111. Tanaka H, Yoshida T, Miyamoto N, Motoike T, Kurosu H, Shibata K, et al. Characterization of a family of endogenous neuropeptide ligands for the G protein-coupled receptors GPR7 and GPR8. Proc Natl Acad Sci USA 2003;100:6251-6.
112. Liu C, Chen J, Sutton S, Roland B, Kuei C, Farmer N, et al. Identification of relaxin-3/ INSL7 as a ligand for GPCR142. J Biol Chem 2003;278:50765-70.
113. Liu C, Eriste E, Sutton S, Chen J, Roland B, Kuei C, et al. Identification of relaxin-3/INSL7 as an endogenous ligand for the orphan G-protein- coupled receptor GPCR135. J Biol Chem 2003;278:50754-64.
114. He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004;429:188-93.
115. Cherezov V, Rosenbaum DM, Hason MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007;318:1258-64.
116. Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human beta2 adrenergic G-Protein-Coupled Receptor. Nature 2007;450:383-7.
117. Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, et al. A specific cholesterol binding site is established by the 2.8 A structure of the human beta(2)-adrenergic receptor. Structure 2008;16:897-905.
118. Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane Jr, et al. The 2.6 angstrom crystal stricture of a human A2A adenosine receptor bound to an antagonist. Science 2008;322:1211-7.
119. Warne A, Serrano-Vega MJ, Baker R, Moukhametzianov R, Edwards PC, Henderson R, et al. Structure of the beta1-adrenergic G protein coupled receptor. Nature 2008;454:486-91.
Received in March, 2007.
Accepted for publication in October, 2008.
Ania de la Nuez Veulens. División de Química Física, Centro de Ingeniería Genética y Biotecnología, CIGB Apartado Postal, 6162, Ciudad de La Habana, Cuba. E-mail: ania.delanuez@cigb.edu.cu

{kind=link}