










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.26 no.2 La Habana Apr.-June 2009
RESEARCH
Transgenic tobacco cells: a permissive system for the assessment of resistance strategies against Tomato yellow leaf curl virus
Células de tabaco transgénico: sistema permisivo para la evaluación de estrategias de resistencia al virus del enrollamiento foliar amarillo del tomate
Alejandro D Fuentes, Pedro L Ramos, Ana I Fernández, Kenia Tiel, Danay Callard, Yadira Sánchez, Merardo Pujol
Plant Virology Laboratory, Plant Department, Center for Genetic Engineering and Biotechnology, CIGB. Ave. 31 / 158 and 190, Cuabanacán, Playa, PO Box 6162, CP 10600, Havana, Cuba
ABSTRACT
Tomato yellow leaf curl virus (TYLCV) is a major threat to tomato production in the tropics and sub-tropics around the world. The application of genetic engineering and pathogen derived resistance mechanisms to obtain tomatoes that are resistant to this pathogen is considered a promising alternative to the current protective practice against the virus. However, the development of transgenic tomato plants that are resistant to the virus is a resourceconsuming and time-consuming procedure, often with unpredictable efficiency, which hinders the evaluation of genetic designs. For this reason an assessment of the strategies against TYLCV replication preceding transgenic tomato development would ensure the protective potential of the candidate transgene. Attempting to circumvent this issue, the present study demonstrated the feasibility of using tobacco cell lines to study the consequences of c1 antisense expression on TYLCV replication. As a result, the transgenic tobacco cell lines were able to produce siRNA that is complementary to the c1 sequence and inhibited TYLCV multiplication, forecasting what would happen in transgenic plants harboring this antiviral strategy.
Keywords: begomovirus, TYLCV, tobacco, post-transcriptional gene silencing, pathogen derived resistance, tomato.
RESUMEN
Tomato yellow leaf curl virus (TYLCV) constituye el mayor peligro para la producción de tomate en el mundo, principalmente en las regiones tropicales y subtropicales. La aplicación de la ingeniería genética y los mecanismos de resistencia derivada del patógeno para obtener tomates resistentes a este patógeno es considerada una alternativa promisoria en la práctica corriente de protección contra este virus. Sin embargo, el desarrollo de plantas transgénicas de tomate resistentes a virus es un procedimiento que consume mucho tiempo y recursos, y en la mayoría de las veces resulta impredecible su eficiencia, lo cual dificulta la evaluación de los diseños antivirales. Por ello la evaluación de las estrategias que inhiban la replicación de TYLCV en un sistema previo a la obtención de plantas de tomate transgénicas aseguraría el potencial protectivo del transgén candidato. En un intento por evadir el proceso de transformación de tomate en el presente trabajo se demostró la factibilidad de utilizar líneas celulares de tabaco para estudiar las consecuencias de la expresión del gen c1 en antisentido sobre la replicación de TYLCV. Como resultado las líneas celulares de tabaco fueron capaces de producir ARN de interferencia, complementarios a la secuencia del gen c1, e inhibieron la multiplicación de TYLCV, previendo lo que sucedería en las plantas transgénicas portadoras de esta estrategia antiviral.
Palabras clave: begomovirus, TYCCU, tabaco, silenciamiento génico post-transcripcional, resistencia derivada del patógeno, tomate.
INTRODUCTION
The tomato yellow leaf curl disease is a major threat to tomato production in tropical and subtropical areas of the world (1). This disease is caused by several closely related begomoviruses, among them the Tomato yellow leaf curl virus (TYLCV), Tomato yellow leaf curl Axarquia virus, Tomato yellow leaf curl China virus, Tomato yellow leaf curl Guandong virus, Tomato yellow leaf curl Kanchanaburi virus, Tomato yellow leaf curl Malaga virus, Tomato yellow leaf curl Mali virus, Tomato yellow leaf curl Sardinia virus and Tomato yellow leaf curl Thailand virus. The use of resistant plants is considered the most reliable and economica-lly practicable strategy to control this whitefly trans-mitted disease (2, 3).
Breeding programs to develop TYLCV resistant varieties have enabled the recognition of plant genes which are involved in the inhibition or attenuation of the symptoms produced by begomoviruses (2, 4-6). However, the transfer of resistance gene from wild tomato species to commercials ones is accompanied by changes in the agronomical properties of recipient cultivars, which could limit the recovery of the commercial standards (6, 7). On the other hand, although the resistant lines produced do not show any symptoms, they are still a source of TYLCV inoculants, since they can not block virus replication. With this resistant phenotype the plants remain in a healthy and optimal physiological state, feeding and increasing the whitefly population. As an alternative to these inconveniences, genetic engineering allows the application of pathogen derived resistance, a mechanism described in 1985 (8) that allows the plants carrying a specific pathogen-derived genomic sequence to withstand viral infection (9). However, plant transformation and regeneration methodologies are still not a routine process for many important tomato cultivars, and transgenic plant production is a resource consuming and time consuming procedure, often with unpredictable efficiency (10), which complicates the evaluation of genetic designs.
The development of simple, reliable and demonstrative systems for assessing virus replication preceding transgenic plant production would facilitate the analysis of the protective potential of a candidate transgene. Certain experimental models have been described, in which suspension cells or protoplasts are inoculated with the genomes of some geminiviruses (11-16). A large part of previous studies were focused on the replication of the bipartite begomoviruses Tomato golden mosaic virus (TGMV), African cassava mosaic virus (ACMV) and Bean golden mosaic virus (BGMV) in tobacco cell lines.
To date, no data is available on the behavior of the monopartite begomovirus TYLCV in replication studies on suspension cells as alternatives, which could simulate conditions for the analysis of inhibition and multiplication, avoiding transgenic plant generation. Here, the tobacco NT1 cell line (17) was assessed as a system, to study TYLCV replication. The research was focused on the inhibition of TYLCV multiplication by generating siRNA derived from the expression of the TYLCV replication associated protein gene (c1) in antisense orientation. As a result, we demonstrated the feasibility of using tobacco NT1 cell line to simulate conditions for TYLCV replication and to forecast what would happen in transgenic plants harboring this antiviral strategy.
MATERIALS AND METHODS
Maintenance of tobacco cell suspension culture
NT1 cell suspension line was maintained as described previously (17), in a liquid Murashige and Skoog (MS) medium (18) supplemented with 2,4-dichlorophenoxyacetic acid (2 mg/L), sub-culturing every week by inoculating 10 mL of the suspension to 100 mL of the fresh medium.
Gene constructions
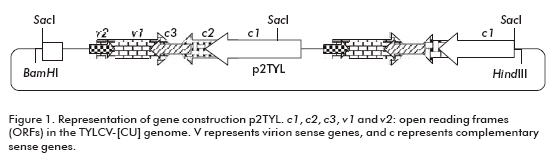
To generate a 2 mer TYLCV-[CU) genomic clone, the plasmid pZTYL (1 mer TYLCV-[CU]) previously obtained in our laboratory at CIGB in Cuba, by cloning the TYLCV-[CU] genome into the pZeroTM-2.1 plasmid (Invitrogen, San Diego, CA), was partially digested with Sac I for 20 min. At the same time a copy of the TYLCV-[CU] genome was generated by a complete digestion with the same enzyme for one hour. The 6.2 kb product of the partial digestion (pZero + TYLCV-[CU]) was treated with alkaline phosphatase and ligated for 4 hours at room temperature to the 2.8 kb fragment of TYLCV-[CU], obtained from a complete digestion (generation of the plasmid pZ2TYL). After transforming Escherichia coli cells, XL1-blue strain, a head to tail clone was selected. The dimer of the TYLCV-[CU] was digested from pZ2TYL by Hind III/BamH I enzymes and ligated into the binary vector pPZP200, previously digested with HindIII/BamHI (generation of the p2TYL plasmid).
The TYLCV-[CU] c1 gene was amplified by polymerase chain reaction (PCR) from the 2 mer TYLCV genomic clone (plasmid p2TYL). Primers were designed to add convenient restriction sites to the end of the amplicons. The c1 gene was amplified with primers A (TCTCTCGAGTTACGCCTTATTGGTTTC) and B (CGCGGATCCATGCCTCGTTTTATTTAAA) (1 min at 94 ºC, 1 min at 46 ºC and 3 min at 72 ºC, in 35 cycles), digested with XhoI-BamHI restriction enzymes and cloned into the pBPU8 plasmid (19) digested with XhoI and BamHI (generation of the plasmid pBPc1antisense). The fragment containing the c1antisense under the 35S promoter and the tNOS terminator was digested from pBPc1antisense with Hind III and cloned into the binary vector pPZP200 digested with Hind III (generation of the plasmid pPZPc1antisense.
Agroinoculation of tomato plants
Tomato plants were grown up to the four to five leaves stage. Under the temperature of 24 ºC and a daylight regime the plants were subjected to a longitudinal incision using a scalpel, at the lower part of the stem. A 100 μL of Agrobacterium tumefaciens suspension, strain C58C1 (pGV2260) (20) harboring the p2TYL plasmid and conditioned for tomato transformation as described previously (10), was inoculated within the injured tissues. The agroinoculated plants were grown for several weeks under the same environmental conditions until the TYLCV replication assays were carried out.
Transformation of tobacco cells
The NT1 cell line was transformed using the gene bombardment system designed by Finer et al. (21). Cells at the exponential growth state (4th day after the sub-culture) were harvested in a number of 5 x 105 units and plated on Whatman #1 discs with the aid of a Buchner funnel. Before the bombardment, discs were incubated in the dark for 6 hours at 24 ºC on the MS medium (18) containing sorbitol and mannitol 0.2 M. Gold particles (1-3 µm) carrying each plasmid were prepared according the procedure described by Sanford (22). Particle mixtures for bombardment contained 50 µL of gold particles (60 mg/mL), 10 mL of the plasmid (3 µg/µL), 50 µL CaCL2 (2.5 M), and 20 µL spermidine (0.1 M). After incubation for 10 min at room temperature, the particles were rinsed in 140 µL of ethanol 95% and resuspended in 25 µL of ethanol 95%. For bombardment, 3 µL of the mixtures were used per shot. After shots, samples were incubated for two days in a liquid NT1 medium. To generate transgenic cell lines, the bombarded discs were placed on a solid NT1 medium (solidified with Phytagel 2 g/L) containing kanamycin 25 mg/L, and 20 days later, antibiotic concentration was adjusted to 150 mg/L. Kanamycin resistant calluses were propagated in the same medium or disaggregated in the liquid NT1 medium for 15-20 days to develop the cell suspension.
Analysis of TYLCV-[CU] replication in tobacco cells
After the first two days of bombardment, total DNA of the NT1 line cells was extracted by grinding in
liquid nitrogen, using 500 µL of the extraction buffer containing urea 7 M, NaCl 5 M, Tris-HCl 1 M pH 8, ethylenediaminetetra-acetic acid (EDTA) 0.5 M, Sarcosyl 0.7%. Extracts were incubated for 30 min at room temperature and shaken periodically. Later, mixtures were treated with phenol/chloroform (v/v) and total DNA was obtained after precipitation in isopropanol.
The DNA digested with the Mung Bean or Mbo I nucleases was analyzed by Southern blot. Briefly, fifteen micrograms of DNA were fractionated in 0.8% agarose gel and transferred onto Hybond N+ membranes (Amersham Pharmacia Biotech, USA) using a TE 80, Vacuum Blotting Unit (Amersham Pharmacia Biotech, USA). DNA was cross-linked to the membrane using an Ultraviolet Crooslinker device (Amersham Pharmacia Biotech, USA). Hybridizations were done at 60 ºC using the [α-32P]dATP-labelled TYLCV c2 gene (capsid protein gene) or the entire viral genome.
Extraction of RNA from transgenic tobacco cells and siRNA detection
For total RNA extraction, 0.5 g of NT1 cells were ground in the presence of liquid N2. After the addition of the extraction buffer (LiCL 0.1 M, Tris-HCL 100 mM pH 8, EDTA 10 mM and sodium dodecyl sulfate (SDS) 1%) and saturated phenol pH 4.4, the mixtures were homogenized by vortex for 30 s and 250 mL of chloroform-isoamylalcohol (24:1, v/v) was added. The water phase was removed after centrifugation and a volume of LiCL 4 M was added. RNAs were allowed to precipitate at -20 ºC and later collected by centrifugation. The pellets were dissolved in water and precipitated in the presence of NaOAc 3 M pH 5.2 and 2 volumes of cold 95% ethanol. Later, the pellets were washed with 75% ethanol and resuspended in water. High molecular weight RNA was discarded after the precipitation of 10 µg of total RNA with polyethylene glycol (MW 8000) 5% and NaCL 0.5 M. Low molecular weight RNAs were precipitated in the presence of 0.1 volume of NaOAc 3 M pH 5.2 and 3 volumes of ethanol 95%. For blotting, both RNAs were denatured at 65 ºC and fractionated in denatured conditions. Fractionation of low molecular weight RNA was performed on 15% polyacrylamide gel containing 7 M urea and transferred to the Hybond N+ membrane (Amersham Pharmacia Biotech, USA) using a TE 80, Vacuum Blotting Unit (Amersham Pharmacia Biotech, USA). In the case of high molecular weight RNA, the fractionation was carried out in 1% agarose gels containing 6.84% formaldehyde. RNAs were crosslinked to the membrane using an ultraviolet Crosslinker device (Amersham Pharmacia Biotech, USA). Hybridizations were done at 50 ºC using the [α-32P]dATP-labelled c1 gene as the probe, prepared by the random primer procedure according to the Prime-a-Gene Labeling System (Promega, USA) instructions.
RESULTS
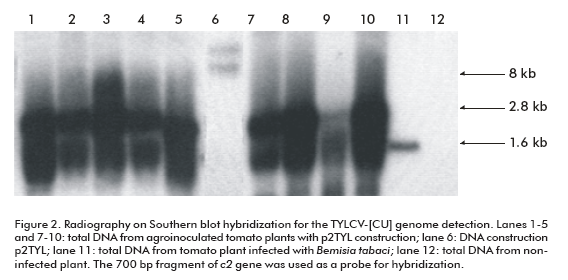
To demonstrate the replicating capacity of the NT1 tobacco cell line for TYLCV, the 2-mer TYLCV-[CU] construction (p2TYL, Figure 1) was transfected into cells applying a particle bombardment procedure. Previously, replication activity of the generated p2TYL was confirmed by agroinoculation into tomato plants. The agroinoculated tomato plants were able to produce double and single strand viral genome DNA corresponding to 2.8 and 1.6 kb respectively, associated to replicative intermediate and viral genome DNA of the new synthesis (lanes 1-5 and 7-10; Figure 2).
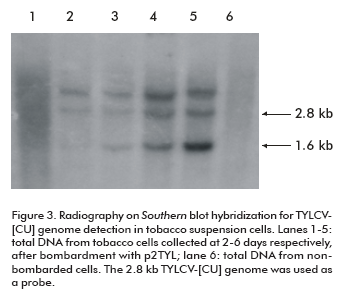
The time course process of viral replication was followed by Southern blot hybridization on total DNA extracted from transfected NT1 cells. As observed on X-ray film, a band of approximately 1.6 kb corresponding to the TYLCV single strand genomic DNA was synthesized after four days of tobacco cell incubation (lanes 3, 4 and 5; Figure 3). Concentrations of TYLCV- [CU] single strand and double strand viral DNA increased daily, and reached the maximum level at the last day of the trial (lane 5; Figure 3).
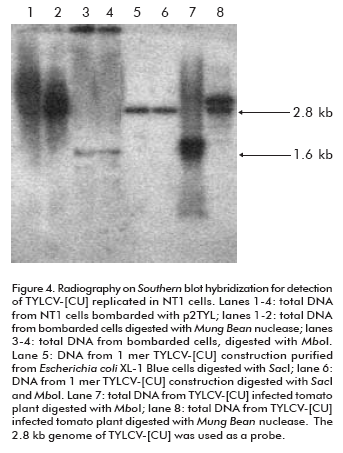
To verify that the 1.6 kb signal corresponds to the TYLCV-[CU] single strand genome, and to evaluate it as an evidence of the new viral genome replicated from the 2 mer construction, total DNAs extracted from bombarded cells were digested with Mung Bean nuclease before the Southern blot hybridization. Since the activity of this enzyme is specific for single strand DNA, the 1.6 Kb signal in treated cell DNA extracts was missing from the autoradiography (lane 1 and 2; Figure 4). In parallel, to confirm the de novo synthesis of the double TYLCV-[CU] strand, total DNAs from inoculated NT1 cells were digested with Mbo I nuclease. Mbo I restriction analysis allows to identify, in a general way, the organism that is the source of a given DNA. Mbo I-recognized DNA sequences harboring a GATC palindrome which is also recognized by Dam methylase in E. coli. The DNA used to transform NT1 tobacco cells was purified from Escherichia coli XL-1 Blue strain which is characterized by Dam methylation (23). Hence, the cloned TYLCV-[CU] genome from E. coli is methylated at MboI restriction sites and could not be digested by this enzyme (lanes 5 and 6; Figure 4). In contrast, the de novo TYLCV-[CU] genome synthesized in NT1 cells is Mbo I susceptible (lanes 3 and 4; Figure 4). Hence, it was demonstrated that the hydrolyzed double strand TYLCV-[CU] genome molecule has been replicated in NT1 tobacco cells.
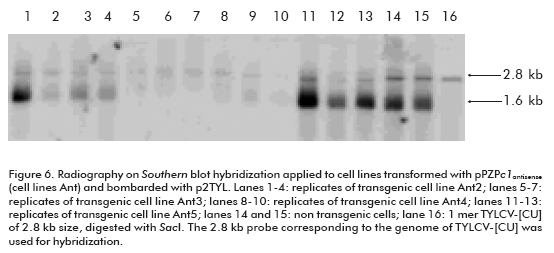
Once the replication of the TYLCV-[CU] genome in the NT1 cell line was established, the next goal of this investigation was to develop transgenic cell lines harboring the antiviral strategy. We chose the construction carrying the c1 antisense sequence (pPZPc1antisense; Figure 5) to induce post-transcriptional gene silencing against TYLCV-[CU] in NT1 cells. The development of transgenic lines was performed by particle bombardment. More than 10 transgenic lines were established in the selective NT1 medium, and all of them were bombarded with p2TYL. Among the transfected transgenic lines there were two that were not able to replicate TYLCV-[CU] (lanes 5-7 and 8-10, corresponding to three replicates of transgenic cell lines Ant3 and Ant4 respectively; Figure 6). Whereas, the rest of the cell lines were able to produce both the double (2.8 kb) and single (1.6 kb) strand viral DNA (lanes 1-4 and 11-13; Figure 6), resembling what had occurred in non transgenic NT1 cells bombarded with the p2TYL construction (lanes 14 and 15; Figure 6). Northern hybridization for the detection of small RNA specific to the c1 sequence evidenced the production of siRNA in the two resistant lines Ant3 and Ant4 (lanes 3 and 4; Figure 7).


DISCUSSION
Tobacco NT1 cell line allows TYLCV replication
The NT1 cell line was selected as a model to analyze the consequences of the TYLCV c1 antisense transgene expression on TYLCV replication due to several practical features: high propagation rate at a low cost, little demand for in vitro manipulation, possibility of being transformed by direct and indirect methods and the short time required to establish transgenic cell lines (22, 24, 25). Probably, the high reproductive capacity of the NT1 cell line favors its competency for its genetic transformation, similar to that suggested for several plant species when they were transformed via Agrobacterium tumefaciens (26-28). As NT1 cells can grow continuously, they should be a good target for systems requiring high transformation frequencies such as the case of our study.
The first step in begomovirus replication consists of an intermediary double stranded DNA (dsDNA) synthesis from a single stranded (SSDNA) genome (29, 30). This process is entirely executed by host enzymes. Afterwards, the start of the rolling cycle replication requires a joint action between viral Rep and REn proteins and cell factors to produce new dsDNAs and ssDNA progenies (31). For several geminiviruses the amount of intermediary dsDNA has been correlated with the occurrence of the S-phase (32-34), and the activation of mitosis and the S-phase entry of the cell is due to the release of the E2F transcription factor. E2F is released through hyperphosphorylation and subsequent inactivation of the retinoblastoma related protein (RBR) that represses it (35). Actually, two geminiviral proteins, Rep and RepA, inactivate the cell RBR, which promotes cell DNA replication necessary for viral genome multiplication (36). Probably the NT1 cell line carries all the necessary elements for the replication of TYLCV by its perennial division state and it could be predicted that no interactions between the geminivirus Rep protein and the repressor of E2F are required. As a supporting fact for this idea other authors have demonstrated the replication of bipartite begomoviruses Tomato golden mosaic virus (TGMV), African cassava mosaic virus (ACMV), Bean golden mosaic virus (BGMV) (13, 14, 16, 37), and monopartite mastrevirus Bean yellow dwarf virus (BeYDV) (38) in tobacco plant cells.
Correspondingly, the NT1 cell line resulted attractive for the establishment of the TYLCV replication model. Therefore, after a two-day incubation period the transfected NT1 cells with the p2TYL construction supported TYLCV replication, which was evidenced by the fact that the dsDNA and ssDNA corresponding signals from the bombarded cells were intensified (figure 3 and figure 4).
As a conclusion we can state that the tobacco NT1 cells contain the necessary elements for TYLCV replication and are therefore an appropriate host for an in vitro model for TYLCV multiplication assays.
Virus challenge: transgenic NT1 tobacco cell line carrying c1 antisense sequence inhibited TYLCV replication
As soon as the NT1 ability for TYLCV replication was demonstrated, we proceeded to assess its capacity to be used in studies on the inhibition of TYLCV multiplication. The cells carrying the c1 antisense construction (Figure 5) were bombarded with 2 mer TYLCV-[CU]. With the aim of detecting the de novo synthesis of the viral genome, total DNA from the bombarded cells was analyzed by Southern hybridization, using the TYLCV genome as the probe.
Two transgenic lines out of ten grown on NT1 solid medium supplemented with kanamycin 150 mg/L showed the capacity to inhibit TYLCV replication; the cell lines Ant3 and Ant4 (Figure 6). This result is evidenced by the absence of autoradiographic signals corresponding to viral dsDNA and ssDNA in comparison to nontransgenic lines.
Previously, an induced post-transcriptional gene silencing (PTGS) against the Rep transcripts aimed to inhibit the ACMV replication in a protoplast system was described (16). For that purpose, the authors introduced specific Small interfering RNA (siRNA) molecules from replication associated gene ac1 sequence into protoplasts of tobacco BY-2 cells, in the presence of ACMV. As a result, partial inhibition of ACMV replication in protoplast, co-transfected with the mixture of siRNA was obtained (16). Although this system has the advantage of giving a rapid demonstration of the suitability of PTGS to inhibit ACMV replication in a protoplast system (in 3-4 days), only a partial effect on ACMV genome multiplication was evidenced in comparison to the transfected samples without siRNA. In addition, ACMV replication still took place, which seems to be controversial since the Rep protein is required for virus replication, although the authors were not able to detect the mRNA specific for Rep by Northern hybridization. In our study the cell transgenic lines Ant3 and Ant4 totally inhibited the TYLCV genome replication (Figure 6), which could prevent the multiplication of TYLCV.
The genetic design to provoke c1 gene silencing was focused on the promotion of double strand RNA (dsRNA) which is expected to induce post-transcriptional gene silencing against the target sequence. To obtain this design the c1 antisense sequence was ligated downstream of the 35S promoter of the Cauliflower mosaic virus (CaMV) and upstream to the nopaline synthetase terminator of the Agrobacterium tumefaciens. This construction, known for its capacity to diminish TYLCV infection in Nicotiana benthamiana transgenic plants (39), should originate complementary transcripts to those produced from the c1 gene of the TYLCV genome. Complementary transcripts could form dsRNA molecules which would constitute the substrate for ARNase type III (Dicer) specific for dsRNA. As a result, fragments of 21 to 25 bp siRNA should be produced (40).
Consistently, transgenic cell lines Ant3 and Ant4 are those producing siRNA complementary to the c1 gene (Figure 7). The inhibition of TYLCV replication in these two lines reflects the capacity of the PTGS mechanism, directed against the Rep, to diminish TYLCV multiplication, and, on the other hand, the capacity of tobacco NT1 cell line to develop the PTGS against the c1 gene.
Nevertheless, transgenic cell lines Ant3 and Ant4 produced siRNA that was complementary to the c1 gene sequence even when not inoculated with TYLCV. The PTGS process in these two lines could be started from a single aberrant strand of RNA which are converted to dsRNA by the action of cell RNA dependent RNA polymerase (RdRP) (41, 42). The siRNA complementary to the c1 gene may be derived from transcripts without the 5´ cap structure or poly A tail at the 3´ end, originated under a high 35S promoter activity (43, 44), as a substrate for RdRP. Also, multiple copies of the c1 antisense sequences may have occurred at high frequencies after the particle bombardment procedure (45, 46) (not demonstrated in the present study) triggering, through an excess of mRNAs, the formation of secondary structures which could be diced into siRNA molecules (47). Consequently, the advantage of the system proposed in the present work consists of the stable production, a priori, of the siRNA molecules complementary to the c1 sequence as the consequence of the transgene integration. Therefore, the synthesis of the Rep protein once the virus is inside the cell is unlikely to occur, which could be explained by the immediate degradation of the viral c1 transcript. In this way, our system closely resembles what could occur in plants carrying this antiviral design. Besides, our model circumvents the lack of the stability evaluation of the silencing effect through successive cell generations in transiently evaluated antiviral designs.
In conclusion, transgenic tobacco NT1 cell lines capable to stably generate siRNA molecules specific for the c1 sequence inhibited the replication of TYLCV. This fact was confirmed later in transgenic tomato lines that harbour double strand RNA forming construction design specific for c1, which became immune with TYLCV inoculation under high whitefly population conditions (48).
The proposed transgenic system of NT1 tobacco cells demonstrated its suitability for replication studies in TYLCV. In addition, the capacity of these cells to generate siRNA and, subsequently, to induce PTGS against viral genes make them an attractive system for studies on viral functions.
ACKNOWLEDGMENTS
This work was funded by the State Council of the Republic of Cuba.
REFERENCES
1. Polston JE, Anderson PK. The emergence of whitefly-transmitted geminiviruses in tomato in the western hemisphere. Plant Dis 1997;81:1358-69.
2. Friedmann M, Lapidot M, Cohen S, Pilowsky M. A novel source of resistance to Tomato Yellow Leaf Curl Virus exhibiting a symptomless reaction to viral Infection. J Amer Soc Hort Sci 1998;123:1004-7.
3. Lapidot M, Friedmann M. Breeding for resistance to whitefly-transmitted geminiviruses. Ann Appl Biol 2002;140(2):109-27.
4. Kasrawi M. Inheritance of resistance to tomato yellow leaf curl virus (TYLCV) in Lycopersicon pimpinellifolium. Plant Dis 1989;73:435-7.
5. Picó B, Diez MJ, Nuez F. Viral diseases causing the greatest economic losses to the tomato crop. II. The tomato yellow leaf curl virus-α review. Scientia Horticulturae 1996;67:151-196.
6. Vidavsky F, Czosnek H. Tomato breeding lines immune and tolerant to tomato yellow leaf curl virus (TYLCV) issued from Lycopersicon hirsutum. Phytophatology 1998;88:910-914.
7. Vidavsky F, Leviatov S, Milo J, Rabinowitch H, Kedar N, Czosnek H. Response of tolerant breeding lines of tomato Lycopersicon esculentum, originating from three different sources L. peruvianum, L. pimpinellifolium and L. chilense to early controlled inoculation by Tomato yellow leaf curl virus (TYLCV). Plant Breeding 1998;117:165-9.
8. Sanford J, Johnston S. The concept of parasite-derived resistance: deriving resistance genes from the parasite´s own genome. J Theor Biol 1985;113:395-405.
9. Beachy RN. Mechanisms and applications of pathogen-derived resistance in transgenic plants. Curr Opin Biotechnol 1997;8:215-20.
10. Fuentes A, Ramos P, Sánchez Y, Callard D, Ferreira A, Tiel K et al. A transformation procedure for recalcitrant tomato by addressing transgenic plant-recovery limiting factors. Biotechnol J 2008;3:1088-93.
11. Fontes EP, Eagle PA, Sipe PS, Luckow VA, Hanley-Bowdoin L. Interaction between a geminivirus replication protein and origin DNA is essential for viral replication. J Biol Chem 1994;269(11):8459-65.
12. Hanson SF, Maxwell DP. trans-Dominant inhibition of geminiviral DNA replication by bean golden mosaic geminivirus rep gene mutants. Phytopathology 1999;89:480-6.
13. Ha C, Coombs S, Revill P, Harding R, Vu M, Dale J. Corchorus yellow vein virus, a New World geminivirus from the Old World. J Gen Virol 2006;87:997-1003.
14. Chatterji A, Beachy RN, Fauquet CM. Expression of the oligomerization domain of the replication-associated protein (Rep) of Tomato leaf curl New Delhi virus interferes with DNA accumulation of heterologous geminiviruses. J Biol Chem 2001;276(27):25631-8.
15. Hou YM, Paplomatas EJ, Gilbertson RL. Host adaptation and replication properties of two bipartite geminiviruses and their pseudorecombinants. Mol Plant Microbe Interact 1998;11(3):208-17.
16. Vanitharani R, Chellappan P, Fauquet CM. Short interfering RNA-mediated interference of gene expression and viral DNA accumulation in cultured plant cells. Proc Natl Acad Sci USA 2003;100(16):9632-6.
17. Paszty C, Lurquin P. Improved plant protoplast plating/selection technique for quantitation of transformation frequencies. BioTechniques 1987;5:716-8.
18. Murashige T, Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant 1962;15:473-97.
19. Coego A, Vázques R, Alfonso R, Coll Y, Pujol M, Ménendez E, et al. Effect of promoter-stimulatory element combination on transient reporter gene expression in tobacco protoplast using PEG-treatment. Biotecnol Apl 1996;13(2):147.
20. Deblaere R, Bytebier B, DeGreve H, Deboeck F, Schell J, Van Montagu M, et al. Efficient octopine Ti plasmid vectors for Agrobacterium-mediated gene transfer to plants. Nucleic Acids Res 1985;13:4777-88.
21. Finer JJ, Vain P, Jones MW, McMullen MD. Development of the particle inflow gun for DNA delivery to plant cells. Plant Cell Report 1992;11:323-8.
22. Sanford JC, Smith FD, Russell JA. Optimizing the biolistic process for different biological applications. Methods Enzymol 1993;217:483-509.
23. Bullock WO, Fernandez JM, Short JM. XL1-Blue, high efficiency plasmid transforming recA Escherichia coli strain with beta-galatosidase selection. BioTechniques 1987;5:376-378.
24. Fuentes A, Ramos P, Ayra C, Rodríguez M, Ramírez N, Pujol M. Development of a highly efficient system for assessing recombinant gene expression in plant cell suspensions via Agrobacterium tumefaciens transformation. Biotechnol Appl Biochem 2004;39:355-61.
25. Gynheung A. High efficiency transformation of cultured tobacco cells. Plant Physiology 1985;79:568-70.
26. Lai Y, Chen L. Flow cytometric analysis of nuclear cell cycle phases in relation to plant regeneration in Petunia hybrida. J Genet Mol Biol 2002;13:13-20.
27. Peña L, Pérez R, Cervera M, Juárez J, Navarro L. Early events in Agrobacterium-mediated genetic transformation of citrus explants. Ann Botany 2004;94:67-74.
28. Villemont E, Dubois F, Sangwan R, Vasseur G, Bourgeois Y, Sangwan-Norreel B. Role of the host cell cycle in the Agrobacterium- mediated genetic transformation of Petunia: evidence of an S-phase control mechanism for T-DNA transfer. Planta 1997;201:160-172.
29. Kammann M, Schalk HJ, Matzeit V, Schaefer S, Schell J, Gronenborn B. DNA replication of wheat dwarf virus, a geminivirus, requires two cis-acting signals. Virology 1991;184(2):786-90.
30. Saunders K, Lucy A, Stanley J. RNA-primed complementary-sense DNA synthesis of the geminivirus African cassava mosaic virus. Nucleic Acids Res 1992;20(23):6311-5.
31. Stanley J. Analysis of African cassava mosaic virus recombinants suggests strand nicking occurs within the conserved nonanucleotide motif during the initiation of rolling circle DNA replication. Virology 1995;206(1):707-12.
32. Accotto GP, Mullineaux PM, Brown SC, Marie D. Digitaria streak geminivirus replicative forms are abundant in S-phase nuclei of infected cells. Virology 1993;195(1):257-9.
33. Grafi G, Burnett RJ, Helentjaris T, Larkins BA, DeCaprio JA, Sellers WR, et al. A maize cDNA encoding a member of the retinoblastoma protein family: involvement in endoreduplication. Proc Natl Acad Sci USA 1996;93(17):8962-7.
34. Xie Q, Sanz-Burgos AP, Hannon GJ, Gutierrez C. Plant cells contain a novel member of the retinoblastoma family of growth regulatory proteins. EMBO J 1996;15(18):4900-8.
35. de Jager S, Maughan S, Dewitte W, Scofield S, Murray A. The developmental context of cell-cycle control in plants. Semin Cell Dev Biol 2005;16:385-96.
36. Gutierrez C. DNA replication and cell cycle in plants: learning from geminiviruses. EMBO J 2000;19(5):792-799.
37. Orozco BM, Hanley-Bowdoin L. Conserved sequence and structural motifs contribute to the DNA binding and cleavage activities of a geminivirus replication protein. J Biol Chem 1998;273(38):24448-56.
38. Mor TS, Moon YS, Palmer KE, Mason HS. Geminivirus vectors for high-level expression of foreign proteins in plant cells. Biotechnol Bioeng 2003;81(4):430-7.
39. Bendahmane M, Gronenborn B. Engineering resistance against tomato yellow leaf curl virus (TYLCV) using antisense RNA. Plant Mol Biol 1997;33(2):351-7.
40. Aravin A, Tuschl T. Identification and characterization of small RNAs involved in RNA silencing. FEBS Lett 2005;579:5830-40.
41. Ahlquist P. RNA-dependent RNA polymerases, viruses, and RNA silencing. Science 2002;296:1270-3.
42. Dalmay T. An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell 2000;101:543-53.
43. Baulcombe D. RNA silencing in plants. Nature 2004;431:356-363.
44. Luo Z, Chen Z. Improperly terminated, unpolyadenylated mRNA of sense transgenes is targeted by RDR6-mediated RNA silencing in Arabidopsis. Plant Cell 2007;19:943-958.
45. Emani C, Sunilkumar G, Rathore K. Transgene silencing and reactivation in sorghum. Plant Sci 2002;162:181-2.
46. Voinnet O, Vain P, Angell S, Baulcombe DC. Systemic spread of sequence-specific transgene RNA degradation in plants is initiated by localized introduction of ectopic promoterless DNA. Cell 1998;95(2):177-87.
47. Vanitharani R, Chellappan P, Fauquet C. Geminivirus and RNA silencing. Trends Plant Sci 2005;10(3):144-51.
48. Fuentes A, Ramos P, Fiallo E, Callard D, Sánchez Y, Peral R, et al. Intron-hairpin RNA derived from replication associated protein C1 gene confers immunity to Tomato yellow leaf curl virus infection in transgenic tomato plants. Transgenic Res 2006;15:291-304.
Received in March, 2009.
Accepted for publication in June, 2009.
Alejandro D Fuentes. Plant Virology Laboratory, Plant Department, Center for Genetic Engineering and Biotechnology, CIGB Ave. 31 / 158 and 190, Cubanacán, Playa, PO Box 6162, CP 10600, Havana, Cuba. E-mail: alejandro.fuentes@cigb.edu.cu
