










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.28 no.1 La Habana Jan.-Mar. 2011
REVIEW
Hepatitis C virus and lipid metabolism: their implications in vaccine development and treatment
Virus de la hepatitis C y metabolismo lipídico: implicaciones para el desarrollo de vacunas y tratamientos
Santiago Dueñas-Carrera
Hepatitis C Department, Center for Genetic Engineering and Biotechnology, CIGB Ave 31 / 158 and 190, ZP 10600, Havana, Cuba
ABSTRACT
The hepatitis C virus (HCV) infects over 170 million people worldwide and is a leading cause of chronic hepatitis and severe forms of liver damage as cirrhosis and hepatocellular carcinoma. There is no vaccine available against this pathogen and the current therapeutic option, based on the combination of pegylated interferon plus Ribavirin, is expensive, produces undesirable side effects, and is effective in approximately half of the patients treated. HCV establishes a complex and not completely understood interaction with the host. In addition to its variability and interference with the immune system function, the HCV life cycle is closely associated with lipid metabolism and this relationship contributes to viral persistence. The present review analyzes the current state of the art in this association and the disturbances generated, mainly expressed as intracellular lipid accumulation in hepatocytes and increased oxidative stress with negative consequences in the immune response. Moreover, the potential impact on the development of vaccines and more effective therapeutic interventions against this virus, in the context of the disorders in lipid metabolism, is discussed. Finally, perspectives for rational intervention, taking into account the dependence of HCV to lipid metabolism, and potential targets, are evaluated.
Keywords: HCV, vaccine, VLDL, lipid, therapy.
RESUMEN
El virus de la hepatitis C (VHC) infecta a más de 170 millones de personas globalmente y es la causa principal de hepatitis crónica y formas graves de daño hepático, como cirrosis y carcinoma hepatocelular. No existe vacuna disponible contra este patógeno y la terapia actual que se basa en la combinación de interferón pegilado más Rivabirina provoca efectos secundarios y solo es efectiva en aproximadamente la mitad de los pacientes tratados. El VHC establece una compleja interacción con el hospedero que aún no ha sido completamente caracterizada. El ciclo de vida del VHC se relaciona estrechamente con el metabolismo lipídico, lo que junto a su variabilidad genética e interferencia con el funcionamiento del sistema inmune contribuye a la persistencia viral. En esta revisión se analiza el estado del arte de tal interacción, así como las alteraciones que provoca, fundamentalmente la acumulación de lípidos en los hepatocitos y el incremento del estrés oxidativo, con la consiguiente afectación a la respuesta inmune. Además, se discute el impacto potencial para el desarrollo de vacunas e intervenciones terapéuticas contra el VHC en el contexto de un metabolismo lipídico alterado. También se abordan las perspectivas para una intervención racional de la infección, teniendo en cuenta la dependencia del VHC en el metabolismo lipídico y los blancos potenciales de tales procedimientos.
Palabras clave: VHC, vacuna, VLDL, lípido, terapia.
INTRODUCTION
Hepatitis C virus (HCV) infection is a worldwide health problem, causing chronic hepatitis in approximately 85% of the cases, with a frequent progress to severe forms of liver damage like cirrhosis and hepatocellular carcinoma (1). HCV is a parenterally transmitted pathogen that frequently induces extra-hepatic disease expressions such as essential mixed cryoglobulinemia and membranoproliferative glomerulonephritis (2, 3). There is no vaccine currently available against this pathogen, and therapeutic treatments, based on pegylated interferon (PegIFN) plus ribavirin are expensive, produce undesirable side effects and are only effective in about one half of the patients (4). Successful response to treatment against HCV infection seems to depend on several factors, involving both, the virus and the host (5-7).
HCV is a single positive strand RNA virus belonging to the Flaviviridae family, hepacivirus genus. The HCV genome encodes a polyprotein co- and post-translationally processed in at least ten viral proteins with different roles in viral pathogenesis (8). Recent advances in HCV cell culture replication have enhanced the knowledge on the HCV life cycle, although the complete picture is yet unknown. However, one thing is clear, the virus and host establish a very complex interaction during infection.
HCV heterogeneity and mutability, as well as a deficient immune response to this pathogen are perhaps the most relevant factors of viral persistence. Six main genotypes have been described for HCV, with important differences in aspects such as response to standard treatment (9). Individuals infected with genotype 1 have the worst response. On the other hand, there is evidence that HCV can replicate in, or at least enter into, cells of the immune system, in addition to the hepatocytes (10). In fact, several immune system mechanisms, both the innate and adaptive responses, related to the potential clearance of HCV infection, are affected in the chronic phase with: increased resistance to interferon, defects in the function of antigen presenting cells and natural killer cells, specific T cell impairment and exhaustion, among others (11, 12). HCV core and E2 proteins have been frequently associated to these effects, although other viral proteins also seem to be involved (13).
On the other hand, although life style is sometimes underestimated, it is relevant for HCV-related disease and treatment outcomes. In addition to alcohol and drug use, patients should avoid the excessive intake of sugar and fat-enriched food. Particularly, liver steatosis, defined as excessive accumulation of lipid in the cytoplasm of hepatocytes, is a frequent histological feature in HCV chronically infected patients (14). In vivo and in vitro studies have indicated that HCV could alter intrahepatic lipid metabolism by affecting lipid synthesis, oxidative stress, lipid peroxidation, insulin resistance and the assembly and secretion of very low density lipoproteins (VLDL) (15-18). The degree of liver steatosis and insulin resistance has been negatively associated to therapy response (19, 20). The strong relationship of HCV and lipid metabolism seems to involve every step of the HCV life cycle bringing up many still unanswered questions. The present review will analyze different aspects of the relationship between HCV and lipid metabolism, and discuss their potential implications in the development of efficacious preventive and therapeutic interventions.
HCV LIFE CYCLE AND LIPIDS
It is well known that HCV circulates in the host as quasispecies, a population of genetically related molecules differing at the nucleotide level (21). In addition, virion particles of different sizes and density have been detected in circulation (22). The existence of particles lacking HCV E1-E2, or completely non-enveloped, has also been described in patients (23). The detection of HCV genomic mutants, mostly lacking the genes encoding envelope glycoproteins, found in both the liver and serum of patients, could be responsible for this type of particle and introduces a further source of variability (23). However, the main cause of differences in size, density and composition in virion particles seems to be related to the different degrees of association (or not) to lipoproteins (22). The role of each virion subpopulation in viral pathogenesis is not completely defined. However, there is a general consensus that most infectious HCV are circulating as Lipo-Viro-Particles (LVPs), lipoprotein-like structures composed of triglyceride-rich lipoproteins bearing apolipoproteins B (ApoB) and E (ApoE), viral nucleocapsids, and envelope glycoproteins (24, 25).
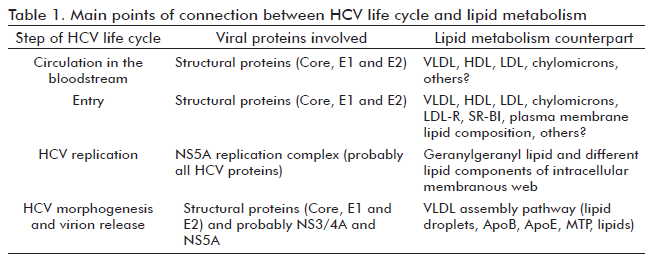
Different pieces of evidence (26) show that HCV exploits lipid metabolism in hepatocytes for entry, replication, assembly and secretion (Table 1). The low density lipoprotein (LDL) receptor, the complement differentiation protein CD81 and the scavenger receptor- class B type I (SR-BI) are molecules involved in early stages of HCV entry to hepatocytes, as attachment factors or co-receptors (27). Hepatocytes can acquire LDL–associated cholesterol in serum through LDL receptor-mediated endocytosis (26). Cells can also acquire cholesterol through SR-BI mediated uptake from high density lipoproteins (HDL), although in humans this pathway is probably not very significant because of the transfer, by cholesteryl ester transfer protein, of cholesterol from HDL to LDL or VLDL (26). HDL has been shown to enhance HCV entry in a process that depends on the lipid transfer function of SR-BI and the presence of apolipoprotein CI (28-30). Interestingly, SR-BI and CD81 are localized in lipid rafts. Remarkably, the amount of CD81 expressed on the cell surface is affected by cellular cholesterol content. The depletion of cholesterol in cells resulted in lower amounts of CD81 located at the plasma membrane, consequently reducing HCV entry (31). Altering the sphingomyelin/ceramide ratio of the plasma membrane can affect HCV entry by also decreasing the cell surface expression of CD81 (32).
Once the virus has entered to the cells, via membrane fusion and endocytosis, the genome is released into the cytoplasm. At this point, cholesterol synthesis in hepatocytes, through the mevalonate pathway, is involved in HCV replication. The product of the mevalonate pathway mainly required for HCV genome replication seems to be geranylgeranyl lipid. It serves as a lipid substrate for protein geranylgeranylation, a post-translational modification that covalently attaches geranylgeranyl to several cellular proteins to facilitate their membrane association (26). Specifically, the geranylgeranylation of the F-box and leucine-rich repeat protein 2 determines its association to the HCV NS5A protein, an interaction regarded as critical for HCV genome replication (26). This last occurs in intracellular membranous webs (8).
HCV core protein accumulates in hepatocytes around lipid droplets, which are intracellular stores of triacylglycerols and cholesteryl esters surrounded by a single layer of phospholipids (33), which can also interact with NS3 and NS5A (34). The HCV core–lipid droplet association seems to be essential for the HCV morphogenesis process, since the envelope and other viral proteins are recruited later in this context (27). Remarkably, lipid droplets are an early step in the assembly of VLDL, the most frequent lipoproteins found in LVPs. In fact, it has been found that the biogenesis pathway of VLDL is involved in HCV morphogenesis with microsomal triacylglycerol transfer protein (MTP), ApoB and ApoE as essential elements in the production of infectious HCV particles (27). Moreover, HCV core expression, at least in transgenic mice, inhibits the activity of MTP and the subsequent secretion of VLDLs (35). This might be a viral strategy to accommodate the production and secretion of VLDL to the rate of HCV virion formation. The final result is the intracellular lipid accumulation.
Indeed, there is strong evidence suggesting that some HCV proteins, particularly the core and NS5A, can induce hepatic steatosis by interfering with intracellular lipid metabolism (36). Two main predominant forces of steatosis have been proposed to coexist in patients with hepatitis C. The first is a metabolic type that is seen mainly in HCV-1 infected patients and is associated with increased body mass index, hyperlipidemia, and insulin resistance. The second is a viral type that may also be developed in the absence of any other steatogenic cofactors and that seems to be directly triggered by the virus through the interference with intracellular lipid metabolism or the induction of insulin resistance (36). These two forces are not mutually excluding but probably synergic in generating hepatic steatosis in hepatitis C patients.
IMPACT ON VACCINE DEVELOPMENT
Different elements support the rationality of generating an effective vaccine against HCV. The most relevant fact is probably that immunity to the virus can be produced since 20-30% of individuals exposed to the virus spontaneously clear the infection and the immune system is critical to this outcome (28). On the other hand, in HCV chronic infections, immune response is not only unable to clear the virus but also seems to contribute to liver damage and extra-hepatic disease expressions (11, 37). Therefore, strategies based on the specific immune modulation, including vaccination strategies are truly promising. However, the generation of vaccines against HCV has become a challenging task.
The resolution of HCV infection requires a complex interplay between innate and adaptative immune responses. In the absence of suitable animal models, only chimpanzees are consistently infected by HCV but major ethics and cost-related reasons have limited its use. Studies using samples from individuals spontaneously eliminating HCV infection have shed light on immunological correlations with HCV clearance. It is generally supported that a strong, multispecific and sustained T-cell response seems to be required for viral clearance. Strategies, generally targeting different HCV antigens, have been evaluated for developing preventive or therapeutic vaccines against this pathogen. In fact, several vaccine candidates have undergone clinical evaluation (38-40). These vaccine candidates were found to be safe and immunogenic (41-43). However, the clinical and virological impact of these vaccine candidates must be demonstrated.
The goal of vaccination against HCV can be seen in three different scenarios: protection or complete clearance of HCV; control of infection avoiding the development of liver damage; the promotion of favorable conditions in the patient for a more effective response after anti-viral treatments. The first setting is ideal but it is undoubtedly the most difficult one to achieve. In fact, the scenario in vaccine development has changed in the last decade, with early studies focused on preventive vaccination and current strategies mainly addressing the therapeutic approach. Hence, most vaccines candidates under clinical evaluation have been designed to elicit cell-mediated immune response. Different factors have led to a decrease in the number of ongoing preventive vaccination studies against HCV. Scientifically, the absence of a complete definition of immunologic parameters correlating with protection and/or the clearance of HCV, and particularly the controversial role of neutralizing antibodies, are probably the most important elements related to this situation. In favor of antibody response, subjects with primary hypogammaglobulinemia showed rapid disease progression and poor response to interferon treatment (44). Moreover, previous studies reported the presence of antibodies specific to E2 HVR in individuals who spontaneously resolved HCV infection (45, 46). However, there is relevant data on the null or delayed induction of neutralizing antibodies in HCV infection (47, 48). Additionally, since at least some neutralizing antibodies are directed towards HVR-I, the induction of this type of response has been involved in selecting viral diversity and a mechanism for viral escape. In other cases, neutralizing antibodies cross-reacting with HCV isolates from different genotypes have been found in chronically infected HCV patients, indicating a high degree of conservation of the targeted epitope (49, 50). Nevertheless, these antibodies, even when induced at high levels, are unable to clear chronic HCV infection (49).
The heterogeneity and mutability of HCV are particularly important for the viral escape from the immune system and persistence. In the light of current knowledge, the association of HCV with lipoproteins poses an additional negative impact for the effective induction and action of neutralizing antibodies. HCV particles may be attached to or incorporated into VLDL during the assembly of the lipoprotein particles and secreted together with VLDL. The nature of the association between HCV and VLDL remains unclear. If HCV hides in the core of VLDL as suggested (22), it makes the virus unique in that the entire virion is not exposed to the serum during circulation. Obviously, if this is the situation, neutralizing antibodies targeted at viral epitopes will not be effective against circulating HCV particles. As previously explained (26), this scenario does not necessarily contradict the observation that the entry of cell culture infectious HCV was inhibited by antibodies targeting viral structural protein E2, since these antibodies may also be included in endocytic vesicles containing HCV. In fact, it has been reported that immunoglobin G can enter clathrin coated pits non-specifically through fluid-phase endocytosis (51). Thus, these antibodies may block HCV entry by binding to the viral structural protein after the virus is released from the lipoprotein particles in endocytic vesicles. Nevertheless, it seems very improbable that HCV may hide completely in VLDL because it requires a dramatical change during viral entry to allow the escape of HCV from VLDL-derived lipoprotein particles, so that the structural protein E2 can interact with its cellular receptors; a step required for HCV entry to the cell. It is more probable that HCV particles associate with VLDL in such a way that some parts of the virion are exposed, including those required in the interaction with cellular receptors. In this case, the HDL-enhancing effect on HCV entry that reduces the sensitivity of HCV to neutralizing antibodies (52, 53) could occur through an increased presence of SR-BI in the cell membrane, thereby reducing exposure time to neutralizing antibodies. In any case, if this partial exposure scenario is correct (according to the nature of the association between HCV and VLDL, which is not completely understood), the viral regions exposed may not always the same in HCV particles. This could be an additional source of viral heterogeneity. Moreover, this might be a viral mechanism to circumstantially disfavor the exposure of relevant immunogenic or neutralizing epitopes.
According to the state of the art on the association between HCV and VLDL, a vaccine against this pathogen designed to generate neutralizing antibodies should target several epitopes at the same time. Additionally, the most relevant epitopes could be conformation-dependent and this conformation may require a lipoprotein context. Moreover, important epitopes could even share regions of both HCV proteins and the VLDL structure itself. In a further degree of complexity, since the composition of VLDL is not always exactly the same, a greater variability is thence expected. Therefore, vaccine candidates involving liposome or lipid moieties in general may be advantageous, although thorough studies are required since there is a risk of inducing or enhancing auto-immune disorders from this manipulation.
On the other hand, in persons that are overweight and/or having baseline liver steatosis and other dysfunctions of lipid metabolism, there were side effects (54, 55) related to oxidative stress, persistent inflammation, disturbance in the signaling cascade of interferon and down-regulation of its receptors, reduction of NKT activity, and other immune-associated disorders, for which preventive vaccination may be less effective. There are two reasons in favor of this: first, vaccine could be less immunogenic in these persons, and second, after being exposed to the virus, infection with HCV may be facilitated by the context of lipid metabolism. Regarding the former issue, overweight and obese persons showed a lower percentage of seroprotection after being immunized with a preventive anti-hepatitis B vaccine (56).
In the present context, vaccine candidates specifically targeting HCV proteins mainly involving undesirable effects in lipid metabolism like nucleocapsid and NS5A could be advantageous. In fact, most vaccine candidates of ongoing clinical trials in HCV-infected individuals target at least one of these antigens. Interestingly, CIGB-230 (a vaccine candidate based on a recombinant HCV core protein co-administered with a plasmid expressing the HCV core, E1 and E2 proteins) elicited predominantly a cell-mediated immune response against the HCV core when administered to HCV genotype 1b patients who were unresponsive to previous treatments with IFN plus ribavirin (38). Liver damage was also reduced in a subset of those patients.
Genetic background has been previously correlated with protection against chronic HCV or clearance after therapy (57). Different regions related to genes involved in immune response such as IL-28B and HLA seem to be critical in this issue (58, 59). HLA is particularly relevant for vaccine design and some currently evaluated vaccine candidates are based on selected HCV epitopes for specific T lymphocytes of relevant HLA (42, 43). Recently, an ApoE genotype has been associated with protection against chronic hepatitis C virus infection (60). Remarkably, ApoE-containing lipoproteins have the ability to modulate key elements of the immune response by either inhibiting or stimulating antigen and mitogen induced T-lymphocyte activation as well as proliferation (61). In fact, ApoE interacts with signals from multiple mitogens including transferrin and interleukin 2 (IL-2), and its impact on the pathology of infectious diseases like hepatitis C, has been linked with its immunomodulatory properties (60).
IMPLICATIONS FOR DRUGS THERAPY
Nowadays, the best therapy is the weekly administration of PegIFN (1.5 μg/kg for PegIFN-alfa-2b or 180 μg/kg for PegIFN-alfa-2a) and the daily ingestion of ribavirin (1000 mg for body weight below 75 kg and 1200 mg for body weight above 75 kg), for six months (in HCV genotypes 2 and 3), or one year (in HCV genotypes 1 and 4). Not all HCV-infected patients are eligible for the treatment. A successful therapy is that of a sustained virological response, established by undetectable HCV RNA levels, six months after the end of the standard treatment. Patients who achieve such a response usually show an improvement in liver histology and clinical outcomes (4).
As previously stated, overweight and liver steatosis has been found to be independent factors for non-response to therapy in patients infected with HCV after the treatment with PegIFN plus ribavirin (19). In this case, the disturbance in the signaling cascade of interferon and down-regulation of its receptors seems to be the main mechanism interfering with the treatment, mainly (although not exclusively) by increasing oxidative stress (54, 62). Interestingly, individuals infected with genotype 3 HCV isolates respond differently (better) to PegIFN plus ribavirin than those infected with genotype 1 isolates (4). This may be due to several causes. Noteworthy, these genotypes, as previously stated, have been described as differently behaving in relation to the predominant hepatic steatosis driving forces and molecules involved in lipid metabolism, mainly due to differences in the HCV core proteins of these genotypes (36). It has been recently demonstrated that the management of dismetabolism, diet and exercise therapy can improve the body mass index, liver histology and, therefore, the response to PegIFN and Ribavirin (63). Since HCV-related alterations of lipid metabolism are supposed to increase with years of infection, the early treatment of patients eligible for therapy with PegIFN plus ribavirin is advised. Therefore, in time, some undesirable co-morbidities caused or enhanced by lipid accumulation and dysfunction, such as heart disease and hypertension, could also worsen the results after therapy or even become contraindications to treatment.
New therapeutic agents specifically targeting essential components of the viral life cycle, such as the HCV NS3/4A serine protease and NS5 RNA-dependent RNA polymerase, are currently in advanced clinical development (64, 65). Interesting results concerning increased sustained virological response, when combined with PegIFN plus ribavirin, has been obtained in clinical practice with some of these molecules (66). However, HCV mutant isolates resistant to these molecules have been described and toxicity is not always low (67). Since all intracellular steps of the HCV life cycle in hepatocytes seem to be associated to the membranous structure and depend on the association of viral proteins with lipid droplets and lipoproteins, molecules targeting polyprotein processing and RNA replication could be also interfered by the lipid environment.
The strong relationship between HCV and lipid metabolism has opened new gates in the search for therapeutic interventions since, for instance, drugs that target cholesterol metabolism may be useful in treating HCV infection. Results show that the treatment of cells with statins (the widely used cholesterol lowering drugs) inhibits HCV RNA replication by depleting geranylgeranyl lipids (68). However, applying statins to treat HCV will require very high doses and would likely cause toxicity in the liver and other organs (26). Other types of drugs used for treating hypercholesterolemia by blocking the assembly and secretion of VLDL, have been found to inhibit the production of HCV particles from infected cells (69). Some of these molecules, i.e. antisense RNA drugs targeting ApoB and several MTP inhibitors, have already been tested in clinical trials (68). Particularly, a long-term treatment with MTP inhibitors led to the toxic accumulation of fat in the liver.
CONCLUSIONS
HCV life cycle and lipid metabolism are connected. Therefore, the rational manipulation of this relationship emerges as a potential strategy for developing preventive and therapeutic interventions against HCV infection. Nevertheless, there must first be a complete definition of the molecular mechanisms governing that relationship. Knowledge on the exact HCV particle architecture and composition is crucial for vaccine development, particularly (but not exclusively) for those strategies designed to elicit neutralizing antibody responses. In the therapeutic setting, the relevance of a multifactorial approach with less-toxic anti-cholesterolemics and immunomodulators, in addition to safer anti-virals, should play a more significant role to eliminate liver and extra-hepatic expressions of HCV infection. Last but not the least, there are the efforts required for population awareness on healthier life styles which, together with scientific achievements for creating effective vaccines or medicines against HCV, will be critical for successful interventions against this pathogen.
ACKNOWLEDGEMENTS
The author would like to acknowledge M.Sc. Yalena Amador for the critical reading of the manuscript.
REFERENCES
- 1. Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25:3834-47.
2. Poanta L, Albu A. Chronic hepatitis C with extrahepatic manifestations. Rom J Intern Med. 2007;45:85-8.
3. Sansonno D, Tucci FA, Ghebrehiwet B, Lauletta G, Peerschke EI, Conteduca V, et al. Role of the receptor for the globular domain of C1q protein in the pathogenesis of hepatitis C virus-related cryoglobulin vascular damage. J Immunol. 2009;183: 6013-20.
4. Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335-74.
5. Aronsohn A, Reau N. Long-term outcomes after treatment with interferon and ribavirin in HCV patients. J Clin Gastroenterol. 2009;43:661-71.
6. Reddy KR, Messinger D, Popescu M, Hadziyannis SJ. Peginterferon alpha-2a (40 kDa) and ribavirin: comparable rates of sustained virological response in sub-sets of older and younger HCV genotype 1 patients. J Viral Hepat. 2009;16:724-31.
7. Maekawa S, Enomoto N. Viral factors influencing the response to the combination therapy of peginterferon plus ribavirin in chronic hepatitis C. J Gastroenterol. 2009;44:1009-15.
8.Brass V, Moradpour D, Blum HE. Molecular virology of hepatitis C virus (HCV): 2006 Update. Int J Med Sci. 2006; 3:29-34.
9. Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962-73.
10. Castillo I, Rodriguez-Inigo E, Bartolome J, de Lucas S, Ortiz-Movilla N, Lopez-Alcorocho JM, et al. Hepatitis C virus replicates in peripheral blood mononuclear cells of patients with occult hepatitis C virus infection. Gut. 2005;54:682-5.
11. Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, Ghrayeb J, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302:659-62.
12. Bode JG, Brenndorfer ED, Haussinger D. Subversion of innate host antiviral strategies by the hepatitis C virus. Arch Biochem Biophys. 2007;462:254-65.
13. Krishnadas DK, Ahn JS, Han J, Kumar R, Agrawal B. Immunomodulation by hepatitis C virus-derived proteins: targeting human dendritic cells by multiple mechanisms. Int Immunol. 2010;22:491-502.
14. Negro F. Hepatitis C virus-induced steatosis: an overview. Dig Dis. 2010;28: 294-9.
15. Patel K, Zekry A, McHutchison JG. Steatosis and chronic hepatitis C virus infection: mechanisms and significance. Clin Liver Dis 2005;9:399-410.
16. Adinolfi LE, Durante-Mangoni E, Zampino R, Ruggiero G. Hepatitis C virus associated steatosis–pathogenic mechanisms and clinical implications. Aliment Pharmacol Ther. 2005;22 Suppl 2:52-5.
17. Lonardo A, Adinolfi LE, Loria P, Carulli N, Ruggiero G, Day CP. Steatosis and hepatitis C virus: mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. 2004;126:586-97.
18. Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabo-lism. Trends Endocrinol Metab. 2010;21: 33-40.
19. Tarantino G, Conca P, Sorrentino P, Ariello M. Metabolic factors involved in the therapeutic response of patients with hepatitis C virus-related chronic hepatitis. J Gastroenterol Hepatol. 2006;21:1266-8.
20. Yaginuma R, Ikejima K, Okumura K, Kon K, Suzuki S, Takei Y, et al. Hepatic steatosis is a predictor of poor response to interferon alpha-2b and ribavirin combination therapy in Japanese patients with chronic hepatitis C. Hepatol Res. 2006; 35:19-25.
21.Kato N. Genome of human hepatitis C virus (HCV): gene organization, sequence diversity, and variation. Microb Comp Genomics. 2000;5:129-51.
22.André P, Perlemuter G, Budkowska A, Bréchot C, Lotteau V. Hepatitis C virus particles and lipoprotein metabolism. Sem Liver Dis. 2005;25:93-104.
23. Pacini L, Graziani R, Bartholomew L, De Francesco R, Paonessa G. Naturally occurring hepatitis C virus subgenomic deletion mutants replicate efficiently in Huh-7 cells and are trans-packaged in vitro to generate infectious defective particles. J Virol. 2009;83:9079-93.
24.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol. 2006;80:2418-28.
25.André P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, et al. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol. 2002;76:6919-28.
26.Ye J. Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus. PLoS Pathog. 2007;3(8):e108.
27. Popescu C-I, Dubuisson J. Role of lipid metabolism in hepatitis C virus assembly and entry. Biol Cell. 2009;102:63-74.
28. Zeisel MB, Fafi-Kremer S, Fofana I, Barth H, Stoll-Keller F, Doffoel M, et al. Neutralizing antibodies in hepatitis C virus infection. World J Gastroenterol. 2007;13:4824-30.
29. Catanese MT, Graziani R, von Hahn T, Moreau M, Huby T, Paonessa G, et al. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J Virol. 2007;81:8063-71.
30. Dreux M, Boson B, Ricard-Blum S, Molle J, Lavillette D, Bartosch B, et al. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J Biol Chem. 2007;282:32357-69.
31. Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol. 2007;81:374-83.
32. Voisset C, Lavie M, Helle F, Op De Beeck A, Bilheu A, Bertrand-Michel J, et al. Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell Microbiol. 2008;10:606-17.
33. Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc Natl Acad Sci USA. 1997;94:1200-5.
34. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089-97.
35. Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chrétien Y, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 2002;16:185-94.
36. Mirandola S, Bowman D, Hussain MM, Alberti A. Hepatic steatosis in hepatitis C is a storage disease due to HCV interaction with microsomal triglyceride transfer protein (MTP). Nutr Metab (Lond). 2010;7:13.
37. Chen CH, Lee CM, Chen CH, Hu TH, Wang JH, Hung CH, et al. Prevalence and clinical relevance of serum autoantibodies in patients with chronic hepatitis C. Chang Gung Med J. 2010;33:258-65.
38. Alvarez-Lajonchere L, Shoukry NH, Gra B, Amador-Cañizares Y, Helle F, Bédard N, et al. Immunogenicity of CIGB-230, a therapeutic DNA vaccine preparation, in HCV-chronically infected individuals in a Phase I clinical trial. J Viral Hepat. 2009;16:156-67.
39.Wedemeyer H, Schuller E, Schlaphoff V, Stauber RE, Wiegand J, Schiefke I, et al. Therapeutic vaccine IC41 as late add-on to standard treatment in patients with chronic hepatitis C. Vaccine. 2009;27:5142-51.
40. Haller AA, Lauer GM, King TH, Kemmler C, Fiolkoski V, Lu Y, et al. Whole recombinant yeast-based immunotherapy induces potent T cell responses targeting HCV NS3 and Core proteins. Vaccine. 2007;25:1452-63.
41. Castellanos M, Cinza Z, Dorta Z, Veliz G, Vega H, Lorenzo I, et al. Immunization with a DNA vaccine candidate in chronic hepatitis C patients is safe, well tolerated and does not impair immune response induction after anti-hepatitis B vaccination. J Gene Med. 2010;12:107-16.
42. Schlaphoff V, Klade CS, Jilma B, Jelovcan SB, Cornberg M, Tauber E, et al. Functional and phenotypic characterization of peptide-vaccine-induced HCV-specific CD8+ T cells in healthy individuals and chronic hepatitis C patients. Vaccine. 2007; 25:6793-806.
43. Yutani S, Yamada A, Yoshida K, Takao Y, Tamura M, Komatsu N, et al. Phase I clinical study of a personalized peptide vaccination for patients infected with hepatitis C virus (HCV) 1b who failed to respond to interferon-based therapy. Vaccine. 2007;25:7429-35.
44. Bjøro K, Frøland SS, Yun Z, Samdal HH, Haaland T. Hepatitis C infection in patients with primary hypogammaglobulinemia after treatment with contaminated immune globulin. N Engl J Med. 1994;331: 1607-11.
45. Zibert A, Meisel H, Kraas W, Schulz A, Jung G, Roggendorf M. Early antibody response against hypervariable region 1 is associated with acute self-limiting infections of hepatitis C virus. Hepatology. 1997;25:1245-9.
46. Dittmann S, Roggendorf M, Dürkop J, Wiese M, Lorbeer B, Deinhardt F. Long-term persistence of hepatitis C virus antibodies in a single source outbreak. J Hepatol. 1991;13:323-7.
47. Stamataki Z, Grove J, Balfe P, McKeating JA. Hepatitis C virus entry and neutralization. Clin Liver Dis. 2008;12:693-12.
48. Pestka JM, Zeisel MB, Bläser E, Schürmann P, Bartosch B, Cosset FL, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci USA. 2007; 104:6025-30.
49. Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, et al. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci USA 2004;101:10149-54.
50. Bartosch B, Bukh J, Meunier JC, Granier C, Engle RE, Blackwelder WC, et al. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc Natl Acad Sci USA 2003; 100:14199-204.
51. Pearse BM. Coated vesicles from human placenta carry ferritin, transferrin, and immunoglobulin G. Proc Natl Acad Sci USA 1982; 79:451-5.
52. Dreux M, Pietschmann T, Granier C, Voisset C, Ricard-Blum S, Mangeot PE, et al. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J Biol Chem. 2006;281:18285-95.
53. Voisset C, Op de Beeck A, Horellou P, Dreux M, Gustot T, Duverlie G, et al. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J Gen Virol. 2006;87:2577-81.
54. El-Zayadi AR. Hepatic steatosis: a benign disease or a silent killer. World J Gastroenterol. 2008;14:4120-6.
55. Kremer M, Thomas E, Milton RJ, Perry AW, van Rooijen N, Wheeler MD, et al. Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis. Hepatology. 2010;51:130-41.
56. Estévez ZC, Betancourt AA, Muzio González V, Baile NF, Silva CV, Bernal FH, et al. Immunogenicity and safety assessment of the Cuban recombinant hepatitis B vaccine in healthy adults. Biologicals. 2007;35:115-22.
57. Nattermann J, Leifeld L, Spengler U. Host genetic factors and treatment of hepatitis C. Curr Mol Pharmacol. 2008;1:171-80.
58. Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338-45.
59. Tseng KC, Chang CK, Chou AL, Hsieh YH, Tseng CA, Lai NS. Prognostic effect of human leukocyte antigen class I and II alleles on chronic hepatitis C patients treated by pegylated interferon-alfa plus ribavirin in Taiwan. Hepatogastroenterology. 2010;57:456-61.
60. Kuhlmann I, Minihane AM, Huebbe P, Nebel A, Rimbach G. Apolipoprotein E genotype and hepatitis C, HIV and herpes simplex disease risk: a literature review. Lipids Health Dis. 2010;9:8.
61. Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622-30.
62. Di Bona D, Cippitelli M, Fionda C, Cammà C, Licata A, Santoni A, et al. Oxidative stress inhibits IFN-alpha-induced antiviral gene expression by blocking the JAK-STAT pathway. J Hepatol. 2006;45:271-9.
63. Testino G, Sumberaz A, Ancarani AO, Borro P, Ravetti G, Ansaldi F, et al. Influence of body mass index, cholesterol, triglycerides and steatosis on pegylated interferon alfa-2a and ribavirin treatment for recurrent hepatitis C in patients transplanted for HCV and alcoholic cirrhosis. Hepatogastroenterology. 2009;56:501-3.
64. Fowell AJ, Nash KL. Telaprevir: a new hope in the treatment of chronic hepatitis C? Adv Ther. 2010;27:512-22.
65. Nyanguile O, Devogelaere B, Vijgen L, Van den Broeck W, Pauwels F, Cummings MD, et al. 1a/1b subtype profiling of nonnucleoside polymerase inhibitors of hepatitis C virus. J Virol. 2010;84:2923-34.
66. Sarrazin C, Rouzier R, Wagner F, Forestier N, Larrey D, Gupta SK, et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology. 2007; 132:1270-8.
67. Curry S, Qiu P, Tong X. Analysis of HCV resistance mutations during combination therapy with protease inhibitor boceprevir and PEG-IFN alpha-2b using TaqMan mismatch amplification mutation assay. J Virol Methods. 2008;153:156-62.
68. Ye J, Wang C, Sumpter R Jr, Brown MS, Goldstein JL, Gale M Jr. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci USA 2003;100:15865-70.
69. Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M Jr, et al. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA 2007;104:5848-53.
Received in August, 2010.
Accepted for publication in February 2011.
Santiago Dueñas-Carrera, Hepatitis C Department, Center for Genetic Engineering and Biotechnology, CIGB Ave 31 / 158 and 190, ZP 10600, Havana, Cuba, E-mail: santiago.duenas@cigb.edu.cu

{kind=link}