










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.28 no.2 La Habana Apr.-June 2011
RESEARCH
Detection of conformational shifts and mutations in exon 8 from the atp7b gene in Cuban Wilson’s disease patients
Detección de cambios conformacionales y mutaciones en el exón 8 del gen atp7b en pacientes cubanos con la enfermedad de Wilson
Yulia Clark1, Teresa Collazo1, Caridad Ruenes2, Elsa García2, Zoe Robaina1, Trini Fragoso3, Gabriel Rodríguez3, Yaixa Piloto1, Georgina Espinosa4, Carlos Maragoto5, Héctor Vera5, Idalmis García2, Lídice Reyes1, Carlos Castañeda2
1Laboratorio de Biología Molecular, Centro Nacional de Genética Médica Ave. 31, esquina 146, Cubanacan, Playa, La Habana, Cuba.
2Instituto Nacional de Gastroenterología Calle 25 e/ H e I, Vedado, La Habana, Cuba.
3Hospital Pediátrico Universitario Pedro Borrás Calzada de Aldabo y E #11117, Alta Habana, Boyeros, La Habana, Cuba.
4Departamento Biología Molecular, Facultad de Biología, Universidad de la Habana, UH Calle 25 e/ I y J, Plaza de la Revolución, La Habana, Cuba.
5Centro Internacional de Restauración Neurológica, CIREN Ave. 25 #15805 e/ 158 y 160, CP 11300, Cubanacan, Playa, La Habana, Cuba.
ABSTRACT
Wilson’s disease is a hereditary disorder of autosomal recessive inheritance that can cause irreversible, potentially lethal lesions to liver and brain. Its molecular cause is the appearance of mutations in the atp7b gene. A total of 379 different disease-producing mutations are currently known, turning the molecular diagnosis of this disorder into a formidable challenge. The present study used single-strand conformational polymorphism for the detection of conformational changes in exon 8 of this gene. Two shifts distinct from the normal allele, denominated b and c, were detected and mapped to mutations L708P and 2304DupC in heterozygosis. Allelic frequencies for these mutations in 72 Cuban Wilson’s disease patients were 2 and 0.7%, respectively.
Keywords: Wilson’s disease, mutation L708P, SSCP.
RESUMEN
La enfermedad de Wilson es un trastorno hereditario que se transmite con un patrón de herencia autosómico recesivo. Puede provocar lesiones irreversibles en el hígado y el cerebro, que pueden llevar a la muerte. Su causa molecular son las mutaciones en el gen atp7b con 379 variantes promotoras de la enfermedad. El diagnóstico molecular es complejo. En este estudio se empleó la técnica de polimorfismo conformacional de simple cadena para la determinación de cambios conformacionales en el exón 8 de ese gen. Se detectaron dos cambios distintos de la variante normal, denominados: b y c, los cuales correspondieron a las mutaciones L708P y 2304DupC en estado heterocigótico, respectivamente. Las frecuencias alélicas de tales mutaciones en 72 pacientes cubanos con la enfermedad de Wilson son de 2 y 0.7%, respectivamente.
Palabras clave: enfermedad de Wilson, mutación L708P, SSCP.
INTRODUCTION
Wilson’s disease (EW, MIM 27790) is a hereditary disorder transmitted in an autosomal recessive pattern. It constitutes a global health problem with a challenging clinical diagnosis. The disorder is characterized by liver damage, ranging from minimal alterations in serum transaminase levels to decompensated cirrhosis and even fulminant hepatitis; in addition, the affected patients often sustain neurological injuries resulting in memory loss, motor impairment, tremors and loss of language skills, among others (1, 2).
Clinical diagnosis of Wilson’s disease is based on the integrated interpretation of a number of clinical and biochemical tests, such as assays for copper in 24-hour urine, copper in serum, ceruloplasmin (although 5 to 15% of patients have normal ceruloplasmin levels), hepatic biopsies (marred by false positives, such as cholestatic syndromes, and false negatives arising from irregular copper distribution) and the presence of Kayser-Fleischer rings, among others (1). The hepatic symptomatology of the disorder may lead to confusion with type 1 hemochromatosis and α-1 antitrypsin.
The estimated worldwide prevalence of Wilson’s disease is 1/30 000. However, prevalence in Cuba has never been measured. atp7b gene (MIM 606882) is found in chromosome 13 at the q14.3-q21 region; in all, 518 variants of this gene have been described, of which 379 are disease-causing mutations (3). More than half of these are nonsense mutations within the transmembrane domains of protein ATP7B and the ATP-binding loop (4-8). Some of the described mutations are also found at splice sites (9).
Large mutational heterogeneity combined with the fact that most molecularly diagnosed patients are compound heterozygotes makes it largely impossible to derive genotype-phenotype correlations for this disease (4-6). In addition, there are reports of individuals homozygotic for mutation H1069Q with differences regarding their clinical symptoms, suggesting the involvement of additional genetic (ATOX1, Apo E) and environmental (including nutritional variability) factors (10).
The molecular diagnosis of Wilson’s disease is a formidable task. Not only there are 21 exons in the atp7b gene, but the number of high frequency mutations is low; varying in number and incidence with the ethnical background and geographical location of every population. Exon 8 is polymorphic; more than 50 disease-causing mutations have been mapped there (3). Mutation L708P affects copper transport and has a frequency of 60% in the Canaries, 16.7% in Brazil, and less than 1% in the United States (11-13); mutation 2304DupC, on the other hand, produces a frameshift and has a frequency of 2.6% in North America, (13). Both have been mapped to exon 8 of the atp7b gene.
Despite its complexities, the molecular diagnosis of this disorder would not only help in the identification of its genetic cause, but serve as confirmation to clinical diagnosis. In addition, it could be used to screen carriers and aid in the identification of anticipatory symptoms, in order to provide early treatment and improve the quality of life of the patient.
The determination of the mutational spectrum of atp7b gene requires an adequate screening technology (4, 14). The detection of single-strand conformational polymorphisms (SSCP) is a technique well-suited for this purpose.
Taking into account that Wilson’s disease has never been molecularly diagnosed in Cuba, the present work is aimed at the search for conformational and mutational changes in exon 8 (polymorphic exon) of the atp7b gene in Cuban patients with this clinical diagnosis.
MATERIALS AND METHODS
The present was a descriptive study, recruiting 72 non-related patients clinically diagnosed with Wilson’s disease. The evaluation was performed by a multidisciplinary team (one geneticist, four gastroenterologists, two neurologists and a biochemist), following inclusion criteria for the diagnosis. Written informed consent by means of a consent form was obtained from each volunteer before inclusion in the study, following the ethical guidelines of the Helsinki declaration. Consent from underage patients, as well as authorization for the selection of this sample, was obtained through their legal tutors. The study posed no risk for the patients, benefitting them instead through genetic counseling. The patients were also briefed on the confidential nature of the research. The study was reviewed and approved by the Ethics Committee of the National Center for Medical Genetics.
Exon 8 from the atp7b gene was selected for the study, due to the number and frequency of Wilson’s disease mutations mapped to this locus for other populations (3).
DNA was extracted by saline precipitation (15) from samples consisting of 10 mL of peripheral blood with EDTA (56 mg/mL) as anticoagulant.
Polymerase chain reaction
The conditions for the amplification of exon 8 by polymerase chain reaction (PCR) were: 100 ng of DNA, 10 pmole/mL each of the oligonucleotides reported by Loudianos et al.: E8 1: 5´ CTA CTT CTT GGC AGC CTT CAC TG 3´, E8 2: 5´ GGA GCA GCT CTT TTC TGA ACC TG 3´ (16), dNTPs at 1 mM (Boehringer), PCR buffer 10X, MgCl2 15 mM, 1 u Taq polymerase (Invitrogen); all in a final volume of 25 mL. The reactions were incubated in an MJ Research thermocycler, following a thermal profile consisting on an initial denaturation of DNA for 4 min at 94 °C, and 35 cycles of denaturation/annealing/extension: 20 s at 94 °C, 30 s at 62 °C, 25 s at 72 °C, respectively.
Single-strand conformational polymorphism (SSCP)
This technique is based on the denaturation of double-stranded DNA products, which are then separated by polyacrylamide gel electrophoresis under non-denaturing conditions. Fragment size, for SSCP to be effective, must be 150 to 300 bp. The power of SSCP resides on the intricate relationship between electrophoretic migration and conformational structure exhibited by single-stranded DNA.
In order to characterize the samples by SSCP, the success of PCR amplification was first verified and then 3.5 µL of a bromophenol blue stop solution (0.05% BPB, 10 mM NaOH, 95% formamide, 20 mM EDTA) were mixed with 1 µL of the amplified product in a final volume of 7 µL, heating the mixture for 5 min at 96 °C and quickly quenching it on ice. The sample was analyzed in a commercially available ready-made acrylamide gel (GeneGel Excel 12.5/24 Kit), run in a Genephor unit at 500 V, 15 W, 15 °C for 2 h. The obtained banding pattern was visualized by silver staining, using the Plus One DNA Silver Staining kit (Amersham Biosciences, 2002). The runs included positive controls for the mutations L708P and 2304DupC.
Detection of mutation L708P
Once the conformational changes of exon 8 were detected, we proceeded to verify the presence of mutation L708P in the patients whose electrophoretic motilities matched that of the L708P positive control. Fifteen microliters of the PCR product were digested with 15 U of Alu I restriction enzyme at 37 °C for 3 h, visualizing the results by electrophoresis in 2% agarose gels at constant voltage (250 V).
Detection of mutation 2304DupC
The presence of the 2304DupC mutation was verified by fluorescent DNA sequencing in an ALF-Express II unit from Amersham Pharmacia Biotech. PCR products were purified with the QIAquick Kit (Qiagen) following instructions from the manufacturer; and the resulting electrophoregrams were analyzed with the ALFwin Sequence Analyser 2.11 and Blast (sequence alignment) software applications.
RESULTS AND DISCUSSION
Studies in Cuba for the detection of mutations of the atp7b gene in local Wilson’s disease patients started in 2008, making an inventory of conformational variants before moving on to the examination of their molecular basis.
The present work detected 3 conformational variants in exon 8 of atp7b through the examination of 72 Cuban patients clinically diagnosed with Wilson’s disease. The identified variants were denominated as a, b and c.
Figure 1 depicts the results of the analysis by 12.5% polyacrylamide gel electrophoresis of exon 8 from 23 out of the 72 patients, together with a positive control. Conformational shifts a, corresponding to the wild type, and conformational shift b, corresponding to mutation L708P in heterozygosis from a control sample, can be observed in the figure 1. Conformer b was detected in 3 out of the 72 clinically diagnosed, non-related patients. SSCP is based on the detection of changes, caused by the appearance of a mutation, on the tridimensional conformation adopted by a single-stranded molecule of DNA, which are reflected in its electrophoretic mobility. Mutation L708P in exon 8 produces, therefore, a conformational change in the DNA strand detectable by electrophoresis, as shown in figure 1.
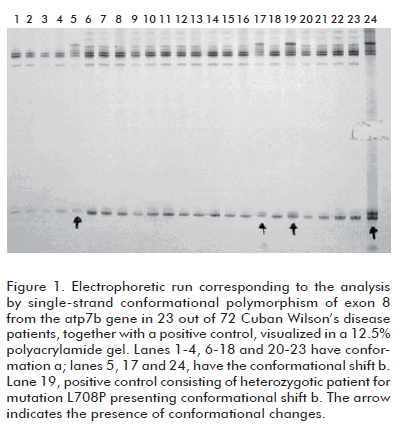
In the samples with conformational change b, the presence of the L708P mutation was confirmed by digestion with the Alu I restriction endonuclease, as the mutation eliminates a recognition site for this enzyme. Patients homozygotic for this mutation produce a single 230 bp band upon digestion, whereas heterozygotes yield 230, 180 and 50 bp bands. Figure 2 demonstrates the presence of L708P in three heterozygotic patients.
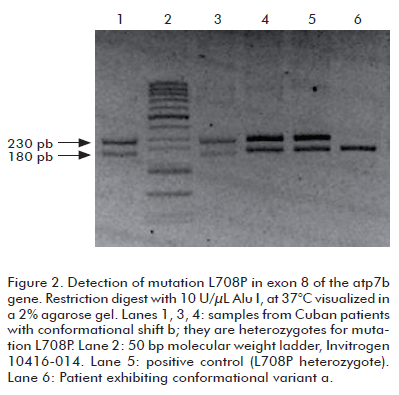
This mutation arises from a thymine to cytosine substitution in position 2123 of the atp7b gene, resulting in a substitution of leucine by proline at residue number 708 in the ATP7B polypeptide. It has been detected in 60% of the cases in the Canaries, making it the most frequent mutation in 24 studied patients (11). However, L708P has not been detected during studies targeting other Spanish populations (4, 17). In Brazil it appears in 16.7% of the cases, being the second most frequent variant in a sample of 60 patients (12); and appearing in less than 1% of 109 patients from 13 states of the USA and Puerto Rico (13). The frequency of this mutation among the 72 analyzed Cuban patients was 2%.
In order to completely characterize the molecular basis of the disorder in the three patients heterozygotic for the L708P mutation, however, it would be necessary to find and catalog the other mutation that, together with the first, produces the symptomatology. Studies are therefore underway to analyze the remaining exons of atp7b gene.
Our work, in any case, provides the country with a fast technology for the molecular diagnosis and determination of mutation L708P in clinically diagnosed patients.
One of the 72 studied samples exhibited conformer C, with the same relative electrophoretic migration as the positive control for mutation 2304DupC in heterozygosis Figure 3. This mutation results from the insertion of a cytosine at position 2304 of atp7b gene, resulting in a frameshift. The presence of this mutation produces a conformational change detectable by SSCP (Figure 3), confirmed by DNA sequencing (Figure 4). This variant was detected at a frequency of 3.3% in the Brazilian sample of 60 patients mentioned above (12) and at 2.6% in the US (13). The frequency in our sample of 72 patients was 0.7%.

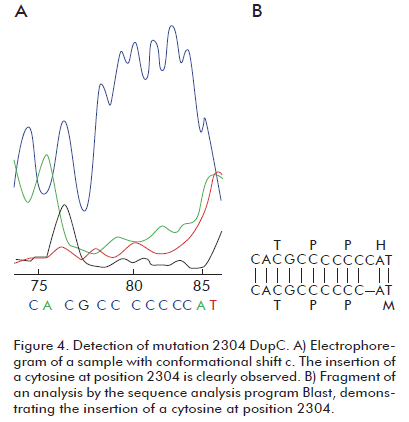
The present is the first study reporting the allelic frequencies for Cuban patients of two mutations in atp7b gene, obtained from the analysis of 72 cases clinically diagnosed with Wilson’s disease. A total of three conformational changes were identified. Sixty-seven of the patients had the normal variant; three had conformer b, corresponding a mutation L708P in heterozygosis, and one had conformational c, corresponding to mutation 2304 DupC.
As mentioned above, the conclusion of the molecular diagnosis of the patients will require identifying mutations and polymorphisms in the remaining exons of atp7b gene, especially those most frequently identified during studies on other populations.
ACKNOWLEDGEMENTS
We thank the patients and their families for agreeing to participate in this study. Theothor Todorov and Georgios Loudianos kindly provided the positive controls and plenty of theoretical discussion; and Antonio Tugores volunteered his expertise in the detection of mutation L708P. We also thank the Cuban Ministry of Public Health.
REFERENCES
1. Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089-111.
2. Durand F. Wilson’s disease: an old disease keeps its old secrets. Eur J Gastroenterol Hepatol. 2007;19(2):97-9.
3. Kenney SM, Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat. 2007; 28(12):1171-7.
4. Margarit E, Bach V, Gómez D, Bruguera M, Jara P, Queralt R, et al. Mutation analysis of Wilson disease in the Spanish population identification of a prevalent substitution and eight novel mutations in the ATP7B gene. Clin Genet. 2005;68(1):61-8.
5. Davies LP, Macintyre G, Cox DW. New mutations in the Wilson disease gene, ATP7B: implications for molecular testing. Genet Test. 2008;12(1):139-45.
6. Abdelghaffar TY, Elsayed SM, Elsobky E, Bochow B, Büttner J, Schmidt H. Mutational analysis of ATP7B gene in Egyptian children with Wilson disease: 12 novel mutations. J Hum Genet. 2008;53(8):681-7.
7. Gromadzka G, Schmidt HH, Genschel J, Bochow B, Rodo M, Tarnacka B, et al. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin Genet. 2005;68(6):524-32.
8. Khaliq A, Majeed A, Asifa S. Spectrum of ATP7B gene mutations in Pakistani Wilson disease patients: A novel mutation is associated with severe hepatic and neurological complication. Int J Biol. 2010; 2(1):117-22.
9. Lovicu M, Lepori MB, Incollu S, Dessì V, Zappu A, Iorio R, et al. RNA analysis of consensus sequence splicing mutations: implications for the diagnosis of Wilson disease. Genet Test Mol Biomarkers. 2009; 13(2):185-91.
10. Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol. 2006;2(9): 482-93.
11. García-Villarreal L, Daniels S, Shaw SH, Cotton D, Galvin M, Geskes J, et al. High prevalence of the very rare Wilson disease gene mutation Leu708Pro in the Island of Gran Canaria (Canary Islands, Spain): a genetic and clinical study. Hepatology. 2000;32(6):1329-36.
12. Deguti MM, Genschel J, Cancado EL, Barbosa ER, Bochow B, Mucenic M, et al. Wilson disease: novel mutations in the ATP7B gene and clinical correlation in Brazilian patients. Hum Mutat. 2004; 23(4):398.
13. Shah AB, Chernov I, Zhang HT, Ross BM, Das K, Lutsenko S, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet. 1997;61:317-28.
14. Lepori MB, Lovicu M, Dessi V, Zappu A, Incollu S, Zancan L, et al. Twenty-four novel mutations in Wilson disease patients of predominantly Italian origin. Genet Test. 2007;11(3):328-32.
15. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
16. Loudianos G, Dessì V, Lovicu M, Angius A, Nurchi A, Sturniolo GC, et al. Further delineation of the molecular pathology of Wilson disease in the Mediterranean population. Hum Mutat. 1998;12(2):89-94.
17. Brage A, Tomé S, García A, Carracedo A, Salas A. Clinical and molecular characterization of Wilson disease in Spanish patients. Hepatol Res. 2007;37(1):18-26.
Received in January, 2010.
Accepted for publication in April, 2011.
Yulia Clark, Laboratorio de Biología Molecular, Centro Nacional de Genética Médica Ave. 31, esquina 146, Cubanacan, Playa, La Habana, Cuba, E-mail: yulia.clark@cngen.sld.cu
