










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.28 no.4 La Habana Sept.-Dec. 2011
RESEARCH
Oxidative damage alters memory consolidation in adult rats
El daño oxidativo afecta la consolidación de la memoria en ratas
Mei-Li Díaz-Hung1, María E González1, Ivette Fernández2, Irenia Horruitiner2, Samuell Piedra1
1Departamento de Inmunoquímica.
2Departamento de Neurofisiología Experimental. Centro Internacional de Restauración Neurológica, CIREN. Avenida 25 no. 15805, CP 11300, La Habana, Cuba.
ABSTRACT
Glutathione is an important cellular antioxidant whose depletion leads to oxidative damage and alters short- and long-term synaptic interactions. Taking into account that the role of this molecule in learning and memory has been poorly studied, the present work evaluates the effect of the administration of L-buthionine sulfoximine (BSO) on glutathione content and markers of cellular oxidative damage (malondialdehyde, superoxide dismutase, glutathione peroxidase). BSO was delivered into the brain by intraventricular injection in the frontal cortex, hippocampus and striatum, evaluating the effect of the resulting glutathione depletion on learning and memory by means of the passive avoidance test, performed 7 days after administration. The results suggest that injecting L-buthionine sulfoximine unbalances the antioxidant enzyme system, resulting in damage to cellular lipid components. In addition, the data suggest the existence of a relationship between oxidative damage originated by glutathione depletion and memory consolidation.
Keywords: oxidative stress, glutathione, L-buthionine sulfoximine, antioxidant enzymes, malondialdehyde, learning and memory.
RESUMEN
El glutatión es un importante antioxidante celular, cuya disminución promueve el daño oxidativo y altera las interacciones sinápticas, a corto y a largo plazo. Su función en el aprendizaje y la memoria ha sido poco estudiada. Se decidió determinar el efecto de la administración del L-butionin sulfoximina sobre el contenido de glutatión, y los indicadores de daño oxidativo celular (malonildialdhehído, superóxido dismutasa, glutatión peroxidasa), a los siete días de la inyección intracerebroventricular, en la corteza frontal, el hipocampo y el cuerpo estriado, así como el efecto de la disminución del glutatión en el aprendizaje y la memoria, mediante la prueba de evitación pasiva. Los resultados sugirieron que el L-butionin sulfoximina induce un desbalance en la actividad enzimática antioxidante, que genera un daño en los componentes lipídicos celulares, y que existe un vínculo entre el daño oxidativo originado por la disminución de glutatión y la consolidación de la memoria.
Palabras clave: estrés oxidativo, glutatión, L-butionin sulfoximina, enzimas antioxidantes, malonildialdehído, aprendizaje y memoria.
INTRODUCTION
Oxidative stress is caused by excess pro-oxidant substances, insufficient antioxidant mechanisms, or both [1]. One of the most important components of cellular antioxidant defense mechanisms is endogen glutathione (GSH), a γ-glutamyl-cysteine-glycine tripeptide present at millimolar quantities in most cell types [2].
GSH protects cells from oxidative damage arising from exposure to excess amounts of exogenous and endogenous electrophilic compounds, contributes to nitric oxide (NO) homeostatic regulation [3] and maintains cytosolic protein thiols in a reduced state [4]. In addition, it is involved in the recruitment, synthesis and release of glutamic acid and γ-aminobutyric acid, as well as the regulation of N-methyl-D-aspartate (NMDA) and non-NMDA glutamatergic receptors [5].
Alterations in the synthesis or steady state levels of GSH have been linked with neuronal death and neurodegenerative diseases such as Parkinson’s and Alzheimer’s disease [6]. Surprisingly little is known, however, about the in vivo role of this antioxidant in cognitive processes.
Considering that memory formation requires changes in synaptic plasticity [7] and taking into account that oxidative stress alters synaptic function [8], our group decided to determine the effect of decreased GSH levels in the activity of the antioxidant enzymes superoxide dismutase (SOD) (E.C. 1.15.1.6) and glutathione peroxidasa (GPx) (E.C. 1.11.1.9), in the levels of malondialdehyde (MDA), an indicator of lipid peroxidation, and in learning and memory.
MATERIALS AND METHODS
Animals
This work employed a total of 120 male, 2 months-old Sprague-Dawley rats supplied by the National Center for the Production of Laboratory Animals (CENPALAB, Mayabeque, Cuba), with an average weight of 250 to 300 grams. They were housed at a density of 5 individuals per box under a temperature of 22 to 24 ºC, with a relative humidity of 60 ± 5% and a photoperiod of 12 h. Bedding was changed twice per week, and water and food were provided ad libitum. All experimental procedures complied with the ethical principles for animal research established by Clark et al. [9] and the Canadian Council for Animal Care (CCAC) [10, 11].
Induction of oxidative stress
Rats of the group receiving the injury were anesthetized with chloral hydrate (420 mg/kg bodyweight, intraperitoneal) and placed in a rodent stereotactic surgery device. An incision was made to expose the cranial Bregma point, from which coordinates corresponding to the ventricle were set: AP -0.8; ML +1.4 and DV -4.0 [12]. Oxidative stress was induced by injecting 7 μL of L-buthionine-(S, R)-sulfoximine (BSO) (10 mM in physiological saline solution) at a rate of 1 µL/min, by means of a 10 µL Hamilton syringe. The group with the sham injury underwent the same surgical procedure, but was administered physiological saline solution instead of BSO. An additional control group was prepared with completely untreated animals.
Preparation of biological samples
Seven days after surgery, the animals were beheaded under deep sedation with chloral hydrate (840 mg/kg bodyweight, intraperitoneal). Their brains were removed and washed with cold physiological saline solution, after which the relevant brain areas (frontal cortex, hippocampus and striatum) from both hemispheres were dissected. Tissue samples were frozen in liquid nitrogen and stored at -70 ºC awaiting further processing.
Quantification of total glutathione
Cytosol fractions were obtained by homogenizing tissue samples in 500 μL of 5% 5-sulfosalicylic acid in a Potter homogenizer (Bioblock Scientific), using 21 strokes at 1000 rpm in an ice-water bath. The obtained samples were centrifuged at 8160 x g for 10 min at a 4 ºC, thus yielding protein-free super-natants. GSH concentrations were determined with the enzymatic recycling method of Tietze [13], incubating 50 μL of a 1.2 mM GSH standard, blank or the corresponding sample with 400 μL of 143 mM sodium phosphate/6.3 mM EDTA/0.24 mM NADPH/0.67 mM 5.5’-dithiobis-2-nitrobenzoic acid at pH 7.5 for 25 min. at 37 ºC in a water bath. This mixture was transferred into a cuvette with 50 μL of glutathione reductase (GRD), 1 U/mL, recording optical density (OD) at 412 nm every 3 seconds for a total period of 1 min in a Shimatzu spectrophotometer. The precipitated mixtures were incubated overnight at room temperature with 500 μL of 1 M NaOH. For protein quantification, they were resuspended by vortexing for several minutes, preparing 1:50 dilutions afterwards.
Determination of superoxide dismutase activity
Brain samples were homogenized in five volumes (w/v) of 1 M Tris/0.25 M sucrose at pH 7.5, using 21 strokes in a Potter homogenizer (Bioblock Scientific) at 1000 rpm in an ice-water bath. The resulting samples were centrifuged for 15 min. at 16 000 x g and 4ºC. SOD activity was measured in the resulting supernatants, using the Marklund method [14]. Sample delipidation was performed by adding 30 µL of chloroform and 50 µL of methanol to every 100 µL of homogeneous compound, followed by vortexing for 1 min. and centrifugation at 2448 x g for 20 min., extracting the supernatants. The assay employed 0.2 M Tris-HCl/2 mM EDTA pH 8.2 buffer. Pyrogallol was prepared at 5% in distilled water and stored protected from light. OD420 nm was recorded every 3 seconds for a total period of 1 min. One unit of enzyme activity was defined as the amount of enzyme inhibiting autoxidation by 50% at 25 ºC.
Determination of glutathione peroxidase enzyme activity
Cytosol fractions were prepared by homogenizing brain samples in 1 mL of 50 mM Tris/0.1 mM EDTA, pH 7.6 in a Potter homogenizer (Bioblock Scientific), using 30 strokes (with intervals of 1 min every 10 strokes) at 1000 rpm in an ice-water bath. The resulting fractions were centrifuged for 15 min at 16 000 x g and 4 ºC, extracting the supernatants and storing them at -20 ºC until further processing.
The assay employed 0.1 M potassium phosphate/1 mM EDTA Na2/2 mM NaN3 pH 7 buffer; solution X (1.25 mM GSH/187.5 µM NADPH/0.3 U/mL GRD in phosphate buffer) and, as substrate, 10 mM CuOOH in 50% ethanol. Fifty microliters of sample (M) or homogenization buffer (B) and 400 μL of solution X were mixed in the cuvettes and incubated for 5 min. at 25 ºC, followed by the addition of 50 μL of CuOOH. OD
Quantification of malondialdehyde
The homogeneous sample was obtained using conditions similar to those employed for the SOD assay. Two-hundred microliters of the sample were vortexed with 400 μL of a 0.67% thiobarbituric acid solution in 0.2 M HCl, then incubated for 15 min in a water bath at 100 °C and centrifuged at 2448 x g for 10 min. at room temperature. The resulting supernatant (200 μLsample + 400 μL reagent) was used for measuring OD535 nm, using blank samples (200 μL H2O + 400 μL reagent) as reference [16].
Quantification of total protein
Total protein was quantified using the Bradford assay [17]. One-hundred microliters of the sample, bovine serum albumin (BSA) standards or the blank were mixed with 2 mL of 0.1 mg/mL Coomassie Brilliant Blue in 8.5% H3PO4 / 0.05% ethanol by vortexing, measuring OD595 nm in the resulting mixtures. Sample absorbances were interpolated in a standard curve prepared with BSA at 0.044, 0.066, 0.88 and 0.132 mg/mL,diluted from a concentrated stock whose concentration was determined based on the extinction coefficient at 280 nm of this molecule (k = 0.68 mL/mg).
Learning and inhibitory memory. Passive avoidance
During the passive avoidance test, animals are placed in a box divided into identically sized lit and dark compartments (each 30 x 30 x 25 cm), communicated through a 7 x 9 cm door, and must suppress their spontaneous preference for darkness in order to avoid a punitive stimulus [18]. At 24 h post-injury, the rats were placed in the lit compartment and were allowed to roam freely through both compartments for 3 min. The training phase started 2 hours later, registering the time of first entry into the dark compartment. After 6 min. of free exploration, they received a 1 mA current (75-80 Hz) pulse of 3 s, after which the door was opened, providing access to the lit compartment. If the rats did not remain in the lit compartment for at least 1 min while avoiding the dark compartment, the stimulus was applied again. At 7 days post-injury, retention was evaluated by reapplying the test without the stimulus, recording the latency time for each crossing between compartments in each phase.
Data processing and statistical analysis
Statistical analyses were performed with the Statistic v6.0 software package. Normal distribution and homogeneity of variance were verified with the Kolmogorov-Smirnov and Levene tests, respectively. Oxidative stress variables were analyzed with a one-way ANOVA followed by Tukey’s test, using instead a non-parametric Kruskal-Wallis test to compare independent groups failing to meet these criteria. Statistical significance was set at p ≤ 0.05.
RESULTS
Biochemical analyses
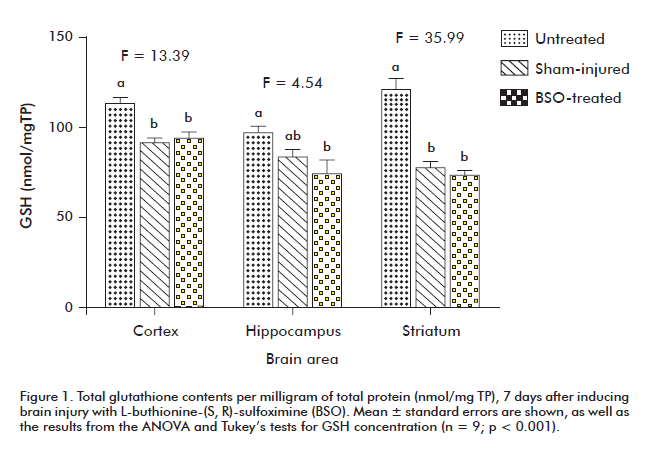
No differences regarding GSH concentration were detected between sham-injured or BSO-treated animals for any of the studied brain areas. However, both groups exhibited statistically significant reductions in the GSH levels of cortex and striatum when compared to untreated animals (p < 0.05) (Figure 1).
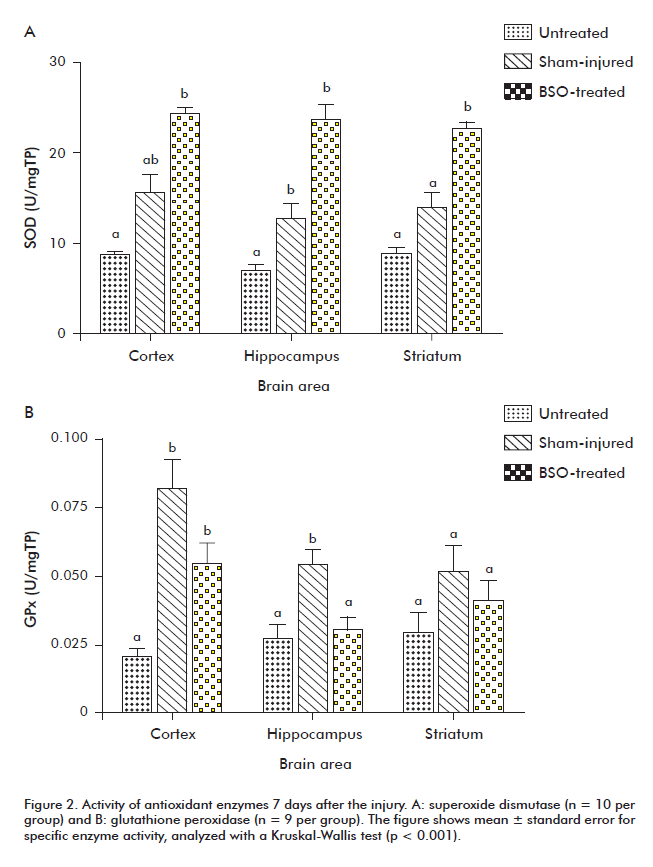
Although SOD activity in BSO-treated animals was significantly higher than in healthy animals, the same comparison, when performed against sham-injured animals, produced statistically significant differences only for striatum. No statistically significant differences were detected between the sham-injured and untreated groups (p < 0.001) (Figure 2A).
GPx activity was highest in the sham-injured group, exhibiting statistically significant differences with the hippocampus of BSO-treated animals and with the cortex and hippocampus of untreated animals (p < 0.01). These differences never reached statistical significance for striatum samples (Figure 2B).
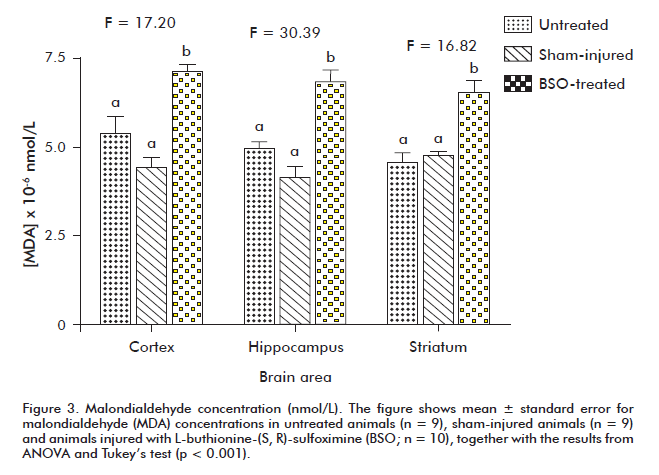
MDA levels exhibited highly significant increases in all the studied brain areas of BSO-treated animals (p < 0.001) (Figure 3).
Behavioral studies
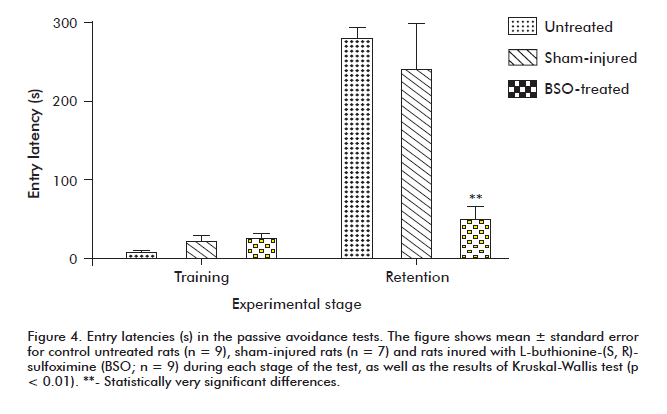
All groups behaved similarly during the training phase in the passive avoidance box. However, when retention was evaluated 7 days later, the control and sham-injured animals exhibited statistically significant (p < 0.01) increases in latency time for entry into the dark compartment when compared with BSO-treated animals (Figure 4).
DISCUSSION
A number of studies have found out that increased oxidative damage correlates positively with early cognitive deterioration in Alzheimer’s disease patients [19]. Normal aging also brings about progressive cognitive impairment, which takes a particularly significant toll in memory-intensive activities. The identification of free radicals as important players in this process has led to the appearance of therapeutic strategies against aging and aging-related pathologies centered on the inhibition or limitation of oxidative reactions involving these agents. Although anti-oxidative therapy has enjoyed growing acceptance, especially in Western countries, hard evidence on its benefits is still lacking due to, among other factors, an insufficient understanding of the oxidative damage mechanisms triggering the process, lack of consensus on what constitute reliable markers of oxidative damage and oxidative status of the patients, and failure to identify a therapeutic window of opportunity [20]. Deeper and more detailed data are required, especially on the intricacies of the GSH system, taking into account its prominent role in the maintenance of oxidative homeostasis in the brain and its link to cognitive function [18].
Different studies in humans, rats and monkeys have demonstrated that the qualitative codification of stimuli takes place in brain structures operating, to a certain extent, as independent memory systems. Such studies have focused mainly on the hippocampus, striatum, amygdala and frontal brain cortex [21].
BSO has received widespread use as inhibitor of γ-glutamylcysteine synthetase during both in vivo and in vitro neurological research. Its effect is, however, transitory, and GSH concentration rebounds back to normal levels by day 7 post-administration [22]. This explains why GSH concentrations do not differ between BSO-treated and sham-injured animals, although both groups still exhibit lower GSH levels than the non-treated group.
Interestingly, MDA levels remained high in the BSO-treated group despite the GSH rebound. This suggests the presence of an oxidative imbalance in this group, supporting the theory that oxidative damage is caused by self-perpetuating mechanisms that outlive the originating event [23]. Decreased GSH concentrations should inhibit the activity of mitochondrial complexes I and II and the production of ATP, which would in turn result in the generation of reactive oxygen species [24]. The latter may diffuse out to other cell compartments where they may cause further damage. Therefore, a compensating response of the antioxidant enzyme system was expected [5].
Although SOD activity remained relatively high in the studied brain areas of BSO-treated individuals when compared with sham-injured animals, this difference reached statistical significance only for striatum samples, suggesting that this region is more susceptible to GSH deficit, as SOD activity is a telltale sign of H2O2 accumulation. GPx uses GSH as electron donor for catalyzing the reduction of H2O2 and other hydroperoxides originating from lipid peroxidation and the metabolism of eicosanoids [25]. Hence, the GSH system is essential for dealing with excess H2O2, protecting biological membranes and other cell components against oxidative damage [26].
That increases in SOD activity are not matched by compensatory increases in GPx activity suggest that the latter is sensitive to the low availability of GSH, specifically in the hippocampus and striatum.
Hydrogen peroxide may oxidize key cysteine residues, altering the structure and function of many proteins [27]. Neurons and glial cells produce NO, a species that can react with O2- anions, thus forming peroxynitrite (ONOO-) and, in a situation where GSH is deficient, nitrosonium (NO+) ions. The latter constitute strongly oxidizing species that can inactivate enzymes such as succinate dehydrogenase, glutamine synthetase, cytochrome P450, the Ca2+-ATPase of the endoplasmic reticulum, Mn2+-SOD and GRD [28].
The low enzyme activity of GPx in BSO-treated animals may arise from oxidative damage of the protein itself resulting, in turn, in accumulated H2O2. Hydrogen peroxide is relatively stable in aqueous solutions, and can easily cross biological membranes [29], triggering sequential reactions of lipid peroxidation and forming aldehydes such as MDA [30]. MDA exhibited increased levels in the studied brain areas of the BSO-treated animals, as expected if a compensatory GPx response is absent, and in agreement with the results obtained by Rougemont et al. after administering BSO to specific brain areas [31].
Some research suggest that the in vitro administration of H2O2 inhibits the activity of NMDA receptors [32]. It has been shown that a 40% deficit in brain GSH is sufficient for decreasing the functionality of NMDA receptors, due to the presence in the latter of cysteine residues sensitive to environmental redox status [33]. Such an effect might well be enhanced by deficits in GPx activity, as observed in the BSO-treated group.
Changes in intracellular Ca2+ concentration mediated by the ion channels of NMDA receptors are essential for activating the protein kinases that modulate, in the long term, the efficacy of synaptic transmission and the levels of protein synthesis required for the maintenance of long-term potentiation, as well as memory consolidation [34].
BSO-treated individuals exhibited low entry latencies and thus, poor retention in the passive avoidance tests. The association between entry into the dark compartment and punitive stimulation took place in seconds, as well as the induction of long-term potentiation [7]. This suggests that these animals are probably unable to maintain the mechanisms that normally guarantee memory consolidation.
Behavioral studies related to oxidative stress have produced disparate results. Some research on the effect of antioxidants on cognitive function suggests a direct involvement of ROS in learning and memory [35, 36]. However, genetic engineering of the β-amyloid peptide has also suggested that oxidative stress is neither necessary nor sufficient to cause memory impairment [37]. GSH deficits have been linked to short-term memory and motivational impairment [18], in addition to favoring oxidative processes that indirectly contribute to cognitive decline [38].
These results suggest that, independently from the triggering event, oxidative damage may persist due to a state of unbalance in antioxidant enzymes that leads to increased lipid peroxidation. The latter condition has a direct effect in biological membranes and may be related to long-term memory impairment in BSO-treated animals.
ACKNOWLEDGEMENTS
The authors would like to acknowledge the contribution of Ariadna Costa Mujica, from the Biochemistry Dept. of the School of Biology of Havana University (Cuba) and Daymara Mercerón Martínez, from the Neurosciences Center (Cuba) to the experimental stage of this work, as well as the help of Lourdes Lorigados, Teresa Serrano and Lissette Blanco, from the International Center for Neurological Restoration (CIREN, Cuba).
REFERENCES
1. Jiménez-Jiménez FJ, Alonso-Navarro H, Ayuso-Peralta L, Jabbour-Wadih T. Estrés oxidativo y enfermedad de Alzheimer. Rev Neurol 2006;42(7):419-27.
2. Drake J, Kanski J, Varadarajan S, Tsoras M, Butterfield DA. Elevation of brain glutathione by gamma-glutamylcysteine ethyl ester protects against peroxynitrite-induced oxidative stress. J Neurosci Res. 2002;(68):776-84.
3. Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic Biol Med. 2004;37(10):1511-26.
4. Chen J, Small-Howard A, Yin A, Berry MJ. The responses of Ht22 cells to oxidative stress induced by buthionine sulfoximine (BSO). BMC Neurosci [internet]. 2005 Feb 15 [cited 2011 Jun 14];6 [about 10 p.]. Available from: http://www.biomedcentral.com/1471-2202/6/10
5. González ME, Fernández I, Bauza JY. Indicadores de estrés oxidativo en cerebros de ratas viejas con déficit cognitivo. Biotecnol Apl. 2007;24:145-50.
6. Chi L, Ke Y, Luo C, Gozal D, Liu R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience. 2007;144(3):991-1003.
7. Izquierdo I, Bevilaqua LR, Rossato JI, Bonini JS, Medina JH, Cammarota M. Different molecular cascades in different sites of the brain control memory consolidation. Trends Neurosci. 2006;29(9):496-505.
8. Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008;45(4):443-52.
9. Clark JD, Gebhart GF, Gonder JC, Keeling ME, Kohn DF. Special Report: The 1996 Guide for the Care and Use of Laboratory Animals. ILAR J. 1997;38(1):41-48.
10. Guidelines for the use of animals in Neuroscience Research. In: Olfert ED, Cross BM, McWilliam AA, editors. Guide to the care and use of experimental animals. Canadian Council on Animal Care. 2nd ed. Ottawa: Bradda Printing Services; 1993. p. 155-62.
11. The use of animals in psychology. In: Olfert ED, Cross BM, McWilliam AA, editors. Guide to the care and use of experimental animals. Canadian Council on Animal Care. 2nd ed. Ottawa: Bradda Printing Services; 1993. p. 163-5.
12. Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 2nd ed. San Diego: Academic Press; 1986.
13. Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997;765(2):283-90.
14. Marklund S, Marlund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974;47(3):469-74.
15. Flohe L, Gunzler WA. Assays of glutathione peroxidase. Methods Enzymol. 1984;105:114-21.
16. Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302-10.
17. Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-54.
18. Cruz-Aguado R, Almaguer-Melian W, Díaz C, Lorigados L, Bergado J. Behavioral and biochemical effects of glutathione depletion in the rat brain. Brain Res Bull. 2001;55:327-33.
19. Markesbery WR, Lovell MA. Damage to lipids, proteins, DNA, and RNA in mild cognitive impairment. Arch Neurol 2007;64:954-6.
20. Fusco D, Colloca G, Lo Monaco MR, Cesari M. Effects of antioxidant supplementation on the aging process. Clin Interv Aging. 2007;2(3):377-87.
21. Rueda-Orozco P, Montes-Rodríguez C, Soria-Gómez E, Herrera-Solís A, Hopkins R, Guzmán K, Próspero-García A, Ruiz-Contreras A, Próspero-García O. Dependencia de los sistemas de memoria al ciclo luz/oscuridad en la expresión de estrategias adaptativas. Primera parte. Salud Mental 2006; 29:19-24.
22. Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol Ther. 1991;51(2):155-94.
23. García R, González M, Fernández I, Castaño Y, Díaz M, Alberti E. Behavioral and oxidative metabolism disorders in a model of transient induced cerebral hipoperfusion in rats. Biotecnol Apl. 2008;25:332-8.
24. Hsu M, Srinivas B, Kumar J, Subramaniant R, Andersen J. Glutathione depletion resulting in selective mitochondrial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: implications for Parkinson’s disease. J Neurochem. 2005;92(5):1091-1103.
25. Liddell JR, Dringen R, Crack PJ, Robinson SR. Glutathione peroxidase 1 and a high cellular glutathione concentration are essential for effective organic hydroperoxide detoxification in astrocytes. Glia. 2006;54(8):873-9.
26. Lv SW, Wang XG, Mu Y, Zang TZ, Ji YT, Liu JQ, et al. A novel dicyclodextrinyl diselenide compound with glutathione peroxidase activity. FEBS J. 2007;274(15):3846-54.
27. Oliveira-Marques V, Marinho HS, Cyrne L, Antunes F. Role of hydrogen peroxide in NF-kappaB activation: from inducer to modulator. Antioxid Redox Signal. 2009;11(9):2223-43.
28. Kuo WN, Kocis JM. Nitration/S-nitrosation of proteins by peroxynitrite-treatment and subsequent modification by glutathione S-transferase and glutathione peroxidase. Mol Cell Biochem. 2002;233(1-2):57-63.
29. Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009;11(6):1349-56.
30. Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269(8):1996-2002.
31. Rougemont M, Do KQ, Castagne V. New model of glutathione deficit during development: Effect on lipid peroxidation in the rat brain. J Neurosci Res. 2002;70(6):774-83.
32. Jiang D, Akopian G, Ho YS, Walsh JP, Andersen JK. Chronic brain oxidation in a glutathione peroxidase knockout mouse model results in increased resistance to induced epileptic seizures. Exp Neurol. 2000; 164(2):257-68.
33. Steullet P, Neijt HC, Cuenod M, Do KQ. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience. 2006;137(3):807-19.
34. Almaguer-Melian W, Cruz-Aguado R, Bergado JA. Synaptic plasticity is impaired in rats with a low glutathione content. Synapse. 2000;38(4):369-74.
35. Li J, Wang C, Zhang JH, Cai JM, Cao YP, Sun XJ. Hydrogen-rich saline improves memory function in a rat model of amyloid-beta-induced Alzheimer’s disease by reduction of oxidative stress. Brain Res. 2010;1328:152-61.
36. Veerendra Kumar MH, Gupta YK. Effect of different extracts of Centella asiatica on cognition and markers of oxidative stress in rats. J Ethnopharmacol. 2002;79(2):253-60.
37. Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med. 2010;48(1):136-44.
38. Shukitt-Hale B, Erat SA, Joseph JA. Spatial learning and memory deficits induced by dopamine administration with decreased glutathione. Free Radic Biol Med. 1998;24(7-8):1149-58.
Received in July, 2011.
Accepted for publication in November, 2011.
Mei-Li Díaz-Hung. Departamento de Inmunoquímica. Centro Internacional de Restauración Neurológica, CIREN. Avenida 25 no. 15805, CP 11300, La Habana, Cuba. E- mail: mldiaz@neuro.ciren.cu.

{kind=link}
{kind=link}
{kind=link}
{kind=link}