










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.29 no.2 La Habana Apr.-June 2012
RESEARCH
Growth Hormone Releasing Peptide 6 (GHRP6) reduces liver fibrosis in CCl4 chronically intoxicated rats
El péptido liberador de la Hormona de Crecimiento-6 (GHRP6) reduce la fibrosis del hígado en ratas intoxicadas crónicamente con CCl4
Jorge Berlanga-Acosta1, Dania Vázquez-Blomquist2, Danay Cibrián1, Yssel Mendoza1, María E Ochagavía3, Jamilet Miranda3, José Suárez4, Yolanda González-Ferrer7, José M Vila7, Angel Abreu7, Dayana Ugarte-Moreno7, Yolanda Cruz7, Ivon Howland7, Rosa Coro-Antich8, Olga S León9, Ricardo Bringas3, Diana García-del Barco1, Karelia Cosme-Díaz4, Daniel Palenzuela2, Julio R Fernández2, Marcelo Nazabal2, Isabel Guillén2, Alberto Cintado2, Lidia Inés2, Ernesto López-Mola5, Gerardo E Guillén-Nieto6
1 Departments of Tissue Repair and Cytoprotection, 2 Genomics, 3 Bioinformatics and 4 Animal Care; 5 Business Development Group and 6 Direction of Biomedical Research. Center for Genetic Engineering and Biotechnology, CIGB. Ave. 31 / 158 and 190, Playa, PO Box 6162, CP 10 600, Havana, Cuba.
7 Center for Medical and Surgical Research.
8 Institute of Neurology and Neurosurgery, INN.
9 Center for Biological Studies, Institute of Pharmacy and Food, University of Havana, UH.
ABSTRACT
Tissue fibrosis is a leading cause of morbidity and mortality. Current treatments for conditions such as hepatic fibrosis have been unsuccessful. The growth hormone relasing peptide 6 (GHRP6) is endowed with cardioprotective actions but its antifibrotic effect had not been anticipated. We examined the GHRP6 ability to prevent and revert liver cirrhosis after induction in Wistar rats by a subcutaneous administration of CCl4. GHRP6 effects were examined after concomitant and delayed administration to toxic respectively. The percentages of hepatic fat, fibrosis, nodularity and septae thickness were histologically and morphometrically determined. Ascitis and portal dilation were judged by ultrasound and serum biochemical profile and oxidative stress parameters determined. Mechanistic involvement of selective gene/proteins was assessed by RT-PCR and immunohistochemistry. Microarrays showed gene expression profiles of GHRP6-treated liver samples on CapitalBio Rat Genome Oligo Array. GHRP6 concomitant intervention prevented in more than 85% parenchymal fibrotic induration (p < 0.0001) and therapeutic administration for only 15 days allowed for 37% fibrotic clearance (p = 0.0004) with more than 30% reduction of septae thickness (p = 0.0011). The 60 days GHRP6 administration scheme produced a 75% reduction of the fibrotic area with more than 60% reduction of nodularity. GHRP6 reduced oxidative damage enhancing the activity of antioxidant enzymes. Vimentin and alpha smooth muscle actin immunodetection profile indicated GHRP6 reduced the number of activated stellate cells. GHRP6 administration reduced fibrogenic factors as TGF-β and CTGF on Kupffer cells. Differentially expressed genes in the microarray experiment indicated GHRP6 modulate the redox balance and parenchymal cells response to injury. These evidences suggest GHRP6 may control the liver’s fibroplastic response.
Keywords: Secretagogue, GHRP6, liver, fibrosis, cirrhosis, carbon tetrachloride, cytoprotection.
RESUMEN
La fibrosis es causa fundamental de morbilidad y mortalidad. Los tratamientos actuales han fracasado. El péptido liberador de la hormona de crecimiento-6 (GHRP6) ejerce efectos cardioprotectores, pero no se ha descrito su acción antifibrótica. Se examinó la propiedad del GHRP6 para prevenir y revertir la cirrosis hepática, luego de su inducción en ratas mediante la administración de CCl4. Se evaluó el porcentaje de grasa hepática, fibrosis, nodularidad y grosor septal mediante estudios histomorfométricos y la ascitis o dilatación portal por ultrasonido. Se determinó el perfil bioquímico y los parámetros de estrés oxidativo en suero, así como la participación de genes y proteínas, mediante reacción en cadena de la polimerasa con transcripción inversa e inmunohistoquímica. Se utilizó un microarreglo de oligonucleótidos del genoma de rata para estudiar el perfil de expresión de los genes inducidos por el GHRP6. La intervención concomitante con GHRP6 previno la induración fibrótica en más del 85% (p < 0.0001). La administración terapéutica durante 15 días permitió la remoción fibrótica del 37% (p = 0.0004), la reducción del grosor septal superó el 30% (p = 0.0011). La administración durante 60 días redujo las áreas fibróticas en 75%, y la reducción de la nodularidad fue de más del 60%. El GHRP6 redujo el daño oxidativo porque aumentó la actividad de las enzimas antioxidantes y las células estrelladas activadas positivas a vimentina y actina alfa de músculo liso. También redujo la expresión de factores fibrogénicos sobre las células Kupffer. El perfil de expresión de genes indicó que el GHRP6 modula el balance redox y la respuesta al daño tisular en las células parenquimales. Estas evidencias sugieren que el GHRP6 pudiera controlar la respuesta fibroplásica del hígado.
Palabras clave: Secretagogo, GHRP6, hígado, fibrosis, cirrosis, tetracloruro de carbono, citoprotección.
INTRODUCTION
Liver fibrosis is the final common pathway of many human hepatic diseases and represents a major source of morbidity and mortality worldwide [1]. In contrast to the traditional view that liver fibrosis is an irreversible disease, recent evidences obtained from animal models and patients indicate that advanced cirrhosis may be ameliorated [2-5].
Hepatic fibrosis is the wound-healing response of the liver to chronic injury [6]. After an acute liver injury, parenchymal cells regenerate and replace the necrotic and/or apoptotic hepatocytes. This process is associated with a controlled inflammatory response and a limited deposition of extracellular matrix (ECM) proteins. If the injury persists, the liver regeneration process fails and the parenchymal hepatocytes are replaced by abundant ECM proteins that disrupt the hepatic architecture by forming cirrhotic nodules. This induces hepatocellular dysfunction and increases intrahepatic resistance to blood flow, which results in liver insufficiency and portal hypertension, respectively [7].
The growth hormone releasing peptide 6 (GHRP6) is a six-amino acids synthetic peptide that belongs to the growth hormone secretagogues (GHS) family. Besides its first described GH-releasing activity [8], mounting evidences substantiate that GHRP6 and their analogs exhibit ever expanding pharmacological effects including cytoprotection [9-13]. We had previously demonstrated that a single GHRP6-prophylactic administration prevented hepatocytes demise in a stringent setting of hepatic ischemia [11]. Whether this GHRP6-induced hepatoprotective effect had impact in liver fibrosis remained unexplored. Nevertheless, serendipitous observations inspired this study, when we observed that rats affected by doxorubicin-induced dilated myocardiopathy and treated with GHRP6 exhibited far less fibrosis in their major parenchymal organs than their saline-treated counterparts.
This work demonstrates for the first time that GHRP6 intervention substantially attenuates the onset of a fibrotic process as well as triggers the regression of cirrhosis in CCl4 chronically intoxicated rats. The intervention appeared to amplify hepatic cells detoxification mechanisms which may ultimately attenuate hepatic stellate cells (HSC) activation and the onset of a fibrogenic program.
MATERIALS AND METHODS
Reagents
The GHRP6 (His-D-Trp-Ala-Trp-D-Phe-Lys-NH2) was purchased from BCN-Peptides (Barcelona, Spain). The product was certified as a sterile, pyrogen-free white powder with 95% purity. For animal administration, fresh solutions were always prepared by diluting the peptide in sterile normal saline solution. CCl4 and mineral oil were purchased from Merck (Darmstadt, Germany). The antibodies anti-Transforming growth factor beta (TGF-β), anti-p53, anti-Cyclin D1 and anti-FasL were purchased from Santa Cruz Biotechnology Inc. (USA). The anti- alpha smooth muscle actin (α-SMA) and anti-Vimentin monoclonal antibodies were purchased from DakoCytomation (Denmark).
Animals
A total of 75 male Wistar rats (250-270 g, 9-10 weeks) were purchased from the National Center for the Production of Laboratory Animals (Havana, Cuba). The rats were maintained in a certified room at the Animal Facility of the Center for Genetic Engineering and Biotechnology (Havana, Cuba), under controlled environmental conditions and with unrestricted animals’ access to food and water. The study protocol was approved by the institutional board on laboratory animals’ welfare.
Fibrosis induction protocol
Liver fibrosis was induced by the subcutaneous injection of CCl4 twice a week (Monday and Friday), for five or seven months. The CCl4 selected dose was 1 mL/kg, diluted as 1:1 proportion with mineral oil before injection [14].
Phases and experimental groups
Seven intact animals received mineral oil for seven months and were terminated once the study was completed (Intact control group). The 68 remaining rats were subjected to the fibrosis induction protocol as mentioned above.
As this study aimed to examine the potential effect of the GHRP6 intervention toward both fibrosis prevention and regression, two experimental blocks were established. The first one, developed during the five initial months, included the concomitant administration of GHRP6 with CCl4 to assess hepatic fibrosis prevention. The second was conducted for the sixth and seventh months while GHRP6 was therapeutically administered to assess its potential in promoting cirrhosis regression.
Experimental groups for the prevention trial
The groups named CCl4 + GHRP6 and CCl4 + Saline received CCl4 as previously described while receiving concomitantly two daily intraperitoneal (i.p.) doses of GHRP6 (400 µg/kg) or normal saline injections, respectively, for five months. Both groups were of 12 rats each and all the animals were autopsied upon concluding the fifth month of the study.
Experimental groups for the regression trial
The 44 remaining rats also received CCl4 as previously described during the five initial months of the study. Afterwards, all these animals were individually subjected to a diagnostic laparotomy (see below). This allowed for allocating the animals into four balanced groups according to their hepatic disease severity.
Two groups of 10 rats each (denominated GHRP6-15d and Saline-15d) received two daily i.p. injections of GHRP6 (dose 400 µg/kg) or normal saline, respectively, for 15 days, starting at the beginning of the sixth month. The CCl4 administration was interrupted during the application of treatments. All these animals were autopsied when the short time intervention was completed.
The last two groups comprised 12 rats each, denominated GHRP6-60d and Saline-60d, respectively. These animals were treated either with GHRP6 or normal saline as described for groups GHRP6-15d and Saline-15d, but for 60 days (sixth and seventh experimental months).The CCl4 administration continued in both groups until the end of the seventh month, when all these rats were finally autopsied.
Hepatic ultrasound
All the animals of the CCl4 + GHRP6 and CCl4 + Saline groups were subjected to a comparative ultrasonic study after five months of concomitant CCl4 and treatments administrations. Animals from GHRP6-60d and Saline-60d groups were comparatively evaluated by ultrasound at the end of the seventh month. The rats from the Intact control group were concurrently evaluated in each case. Ultrasounds were performed with anAloka apparatus (Japan) connected to an 11 MHz transducer in previously anesthetized (Ketamine, 50 mg/kg) animals. The studied parameters included: portal diameter (mm), ascites, parenchymal nodularity, echogenicity increase and liver texture evaluation. Ascites was scored according to the following criteria: 0- no ascites, 1- small quantity of ascites that is only detectable by ultrasound, 2- clinically evident ascites. For parenchymal nodularity a scale from 0 to 3 was used to score the result: 0- no nodules, 1- one or two nodules, 2- a faint multinodularity and 3- multinodular images with large size nodules included. Echogenicity increase was qualitatively graded according to the following scale: 0- no increase, 1- moderate increase and 2- significant increase of parenchymal echogenicity. Liver texture was graded as follows: 0- homogeneous texture; 1- heterogeneous, faintly granulated; and 2- heterogeneous, grossly granulated. The ultrasonic fibrosis index (UFI) was defined as the total sum of the values obtained in these four gradation scales. To avoid a biased judgment all the images from hepatic ultrasound were blindly scored.
Diagnostic study by laparotomy
The 44 rats included in the fibrosis regression phase were subjected to a diagnostic study by laparotomy once concluding the five initial months of CCl4 administration. Briefly, a small abdominal incision was performed to previously anesthetized animals (Ketamine, 50 mg/kg), and by gentle manipulation the whole liver mass was fully exposed for macroscopic inspection, classification and scoring according to the World Health Organization score: macronodular, micronodular, mixed and fat organ gross appearance [15]. Finally, the liver was appropriately returned to its cavity and the small wound was sutured. Balanced groups were made up as judged by the hepatic macroscopic aspect.
Liver histology
For histopathology, three liver fragments from all the animals were harvested during the autopsy, each one from a different hepatic lobe, and were 10% buffered formalin fixed and paraffin embedded. Slides with semi-thin sections (2-3 µm) were prepared and stained with hematoxylin/eosin and Mallory trichrome. All the histological evaluations and morphometric protocols were conducted in a blinded manner. Mallory stained slides were used to assess the cirrhotic nodularity for each animal. For this purpose, the total number of cirrhotic nodules was counted in three equally randomly selected microscopic fields (5 ×) per liver fragment (a total examined area of 10 mm2 by hepatic lobe). The final result was presented as the average of the nodules/mm2 among the three hepatic fragments.
Histomorphometric analysis was conducted using the ImageJ program (NIH, USA). Digital images were captured from the Mallory stained slides in RGB format, with 24 bit true colors and at 3072 × 2304 pixels resolution, through a Carl Zeiss Axiotron microscope (Germany) coupled to a Canon PC1089 camera (Canon, Japan). Fat and fibrosis percentages were assessed for all the animals using images from three equally randomly selected microscopic fields (10 ×) per liver fragment. The final values from the nine captured images were averaged to obtain the representative percentages of fat and fibrosis hepatic covered area for each animal.
In order to determinate the averaged fibrotic septum thickness of each animal, a total of 100 randomly selected microscopic fields (40 ×) was captured by animal, and processed with the calibrated ImageJ software. The thickness was always measured amidst the whole septum length and was reported in microns.
Serum biochemical determinations
Blood samples were collected from the retro-orbital plexus of previously anesthetized rats from the intact control group, on the first experimental day and from all the animals of the CCl4 + GHRP6 and CCl4 + Saline groups after two months of experiment initiation. The final blood samples were harvested by myocardial punction during the autopsies. Serum samples were aliquoted and kept at –20 °C until processing. alanine aminotransferase (ALAT) and aspartate aminotransferase (ASAT) serum activities; as the serum concentration of total proteins (TP), albumin, very low density lipoprotein, cholesterol and triglycerides were assessed in an automatic analyzer Hitachi 747 (Boehringer Mannheim, Germany). Commercial kits and analytical procedures were conducted according to the manufacturer’s instructions.
Hepatic oxidative stress assessment
Liver central lobe fragments collected during the autopsies from all the rats were used to assess the hepatic oxidative stress. The protocols for tissue homogenates and superoxide dismutase (SOD) and catalase enzymes activities were followed as previously described [11]. The lipid peroxidation potential (LPP) and malondialdehyde (MDA) were measured using the Bioxytech LPO-586 commercial kit; while the total hydroperoxide content was assayed by the Bioxytech H2O2-560 commercial kit, both according to manufacturer’s instructions (Bio-Rad Laboratories, Germany). The advanced oxidation protein products (AOPP) content was assessed according to the Witko-Sarsat described technique [16]. All the hepatic biochemical data were adjusted to the total protein concentration determined in the tissue homogenates using a commercial kit (Bio-Rad Laboratories, Germany).
Immunohistochemistry
For immunohistochemistry studies, liver sections (2-3 µm) were mounted on sialinized slides (DAKO, Denmark), heat-treated for antigen exposure, and processed according to the manufacturer’s instructions from DakoCytomation LSABTM+ System-HRP commercial kit. Tissue samples were incubated for 30 minwith: anti-αSMA (1:100), anti-TGF-β (1:250), anti-p53 (1:200), anti-Cyclin D1 (1:100), anti-FasL (1:200) and anti-Vimentin (1:100). Antibodies were diluted in Dako background reducing solution. Immunohistochemistry was accomplished on material retrieved from three representative animals per group purposely selected according to the histopathology judgment. Healthy animals were also included. Slides were counterstained with hematoxilin or light green and were blindly analyzed by two different investigators. The number of Cyclin D1 positively labeled hepatocytes nuclei and FasL positively labeled Kupffer cells within the hepatic parenchyma were quantified in 15 microscopic fields (20 ×), evenly distributed in the three liver fragments collected from GHRP6-15d and Saline-15d groups only. Internal controls included liver sections from healthy intact rats and omission or replacement of the commercial primary antibody by pre-immune isospecies serum. Data are presented as the averaged value of the 45 microscopic fields studied by group (three rats in each one).
Gene expression analyses by semiquantitative RT-PCR
Liver central lobe fragments collected from five randomly selected rats of the Saline-15d, GHRP6-15d and Intact control group were processed to isolate total RNA using TRI Reagent (Sigma, St. Louis, USA). Total RNA was digested with RNase-free DNase I (Epicentre Technologies, USA) according to the manufacturer’s instructions for DNA contaminant removal. Afterward, one microgram of total RNA was reverse transcribed using a commercial available kit (GeneAmp® RNA PCR Core Kit. Applied Biosystems, USA) with an oligo-dT primer. PCR were performed using specific primers and annealing temperatures referred in table 1. Final PCR products were detected in a 1% (w/v) agarose gel and were quantified using the Kodak ID 3.6 software package (Kodak Inc, USA). β-actin was used as housekeeping gene for normalization.
RNA extraction for microarray experiment
Rats in groups GHRP6-60d and Saline-60d were checked for their percentage of fibrosis at the end of the seventh month and fibrosis reduction was then calculated. Total RNA isolation was carried out as for semiquantitative RT-PCR experiments and further purified using a NucleoSpin RNA clean-up kit (Macherey-Nagel, Germany). The quality of the total RNA (i.e., the purity and integrity of the intact RNA) was assessed by Nanodrop 1000 (ThermoScientific, USA) and Bioanalizer Agilent 2100 (Agilent, USA), reporting the concentration, absorbance 260/280 nm ratio of 1.8 or higher, and RNA integrity number equal to or higher than 7, respectively. Five paired samples from GHRP6-60d and Saline-60d that met RNA quality requirements and exhibited a fibrosis reduction superior to 69% were used for the experiment.
Microarray experiment
We chose a reference design with five samples per groups GHRP6-60d and Saline-60d compared to a reference, representing the seven pooled samples from the Intact control group.
The amplification and labeling of mRNA were performed using the CapitalBio cRNA Amplification and Labeling kit according to manufacturing instructions (CapitalBio, Beijing, China). The rat 27 K oligonucleotide microarray comprises 26 962 oligo probes of 70-mer (Capitol-Bio Corporation, Beijing, China) from the Operon Company (Rat Genome Oligo Set, Version 3.0.5). Dual channel microarray hybridization was performed with 70-80 total pmol of Cy3-labeled control sample and Cy5-labeled test samples (from GPHR6-60d and Saline-60d) onto 25 × 75 mm chips. Hybridization and washing of slides were carried out according to the manufacturer instructions (Capitol-Bio Corporation, Beijing, China).The slides were scanned with a confocal LuxScan scanner (CapitalBio Corp, China) and the raw data were extracted using LuxScan™ 3.0 software (CapitalBio Corp). For dual-channels microarray data, the scanning setting for Cy3 and Cy5 channels were balanced. The signals detected from housekeeping and Hex genes, and those detected from the exogenous controls were used as positive controls. Negative controls were at background levels.
Statistical analysis
All the experimental data were initially evaluated for a normal distribution using the Kolmogorov-Smirnov test (p < 0.05). When a normal distribution was established, an unpaired Student’s test was used for comparisons between the groups of prevention phase; while paired Student’s t test was used for the groups of the regression phase. In case of multiple comparisons, the one way ANOVA followed by the Student-Newman- Keuls test was used. The percentage values were compared using the Fisher’s exact test. A value of p < 0.05 was used to indicate a significant difference.
The R Limma package (http://bioinf.wehi.edu.au/limma) was used for preprocessing and differential expression analysis of microarray data [17]. The median average intensity of foreground and background were extracted from the lsr files. A quality criterion [18] was applied to identify low intensity or high background spots assigning weights 0 or 1, which were later used as inputs for limma approach. The normexp+offset method was selected for adaptively adjusting the foreground for the background intensities. The within-arrays normalization was performed with the print-tip loess method using between channels non-differentially expressed controls and for between-array normalization the scale method using all control probes. The moderated paired t-test for each gene was calculated. Genes with p < 0.001 [18] and fold change greater than 1.5 were considered for bioinformatics analysis and biological interpretation.
Bioinformatics analysis of differentially expressed gene
Bioinformatics analysis was performed using, as input, the list of differentially expressed genes in rat (GHRP6-60d vs. Saline-60d) and also the list of their human orthologous in order to take benefit of the functional annotation available on human genes aiming to predict significant biological process involved in the putative GHRP6-mediated antifibrotic mechanisms. Human orthologous for rat genes were identified by searching the Homologene database. Whenever orthologous genes were not found in Homologene, sequence similarity searches of rat’s transcripts against the human RefSeq transcripts were performed with Blastn. The most similar gene, for every rat gene, was chosen as the best Blastn hit (gene with lowest E-value, E-value < 1 × 10-7). To identify putative protein-protein interactions, in which the protein products of the input genes are involved, gene networks were constructed using the Cytoscape’s [19] plugin BisoGenet [20]. All molecular interaction data sources available at BisoGenet were used to generate the gene networks. These networks were enriched with transcription regulation data extracted from the literature. The Cytoscape’s plugin BiNGO [21] was used to identify the Gene Ontology (GO) [22] biological proc[19] plugin BisoGenet [20]. All molecular interaction data sources available at BisoGenet were used to generate the gene networks. These networks were enriched with transcription regulation data extracted from the literature. The Cytoscape’s plugin BiNGO [21] was used to identify the Gene Ontology (GO) [22] biological processes enriched in the lists of input genes. The statistically significant GO processes were determined by using the hypergeometric test and the Benjamini-Hochberg False Discovery Rate (FDR) correction for multiple testing [23] with a threshold of 0.05.
RESULTS
Hepatic ultrasound exploration
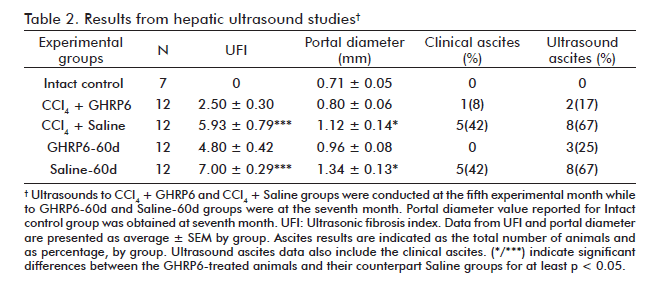
At the fifth month, the comparative ultrasonic study between CCl4 + GHRP6 and CCl4 + Saline groups demonstrated a significant difference (p = 0.0009) between the calculated UFI values, thus suggesting that GHRP6 prevented fibrosis (Table 2). The animals of the GHRP6-60d and Saline-60d groups were also subjected to a comparative study by ultrasound at the seventh month. At this time point, the Saline-60d achieved the largest UFI value registered (Table 2), which was significantly different (p = 0.0005) to that of GHRP6-60d group; thus indicating that a process of fibrosis regression had been set forth.
The portal vein appeared dilated over 60% in the animals from the CCl4 + Saline group at the fifth month, as compared to the Intact control group (p = 0.0052; table 2). In contrast, the averaged portal diameter for the CCl4 + GHRP6 group did not differ from the normal values (p = 0.2938). At the seventh month, once the regression phase concluded, the GHRP6-60d group showed a significant reduction of portal dilation as compared to the Saline-60d group (p = 0.0201).
As shown in table 2, a significant reduction of the animals bearing ascites was detected in the concomitant (p = 0.0361) and the therapeutic GHRP6 for 60 days (p = 0.0373). No ascites was detected for the GHRP6-15d and Saline-15d groups during the short time of therapeutic intervention.
Liver histopathology
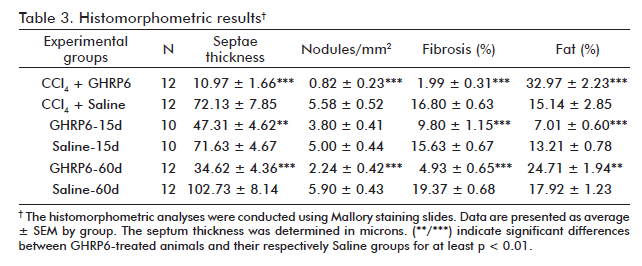
After five months of continuous CCl4 administration, the animals from the CCl4 +Saline group showed dense collagen bundles surrounding cirrhotic nodules (Figure 1A) which represented about 17% of hepatic area covered by fibrosis (Table 3). However, the concomitant intervention with GHRP6 prevented in more than 85% the fibrotic induration (p < 0.0001; figure 1B). In line with this, the CCl4 + GHRP6 group showed far less cirrhotic nodules and averaged lesser septum thickness as compared to the CCl4+Saline group (both p < 0.0001). These findings sustain the ultrasound evidences of GHRP6-mediated anti-fibrotic response. Moreover, the percent of fibrosis, the number of cirrhotic nodules, and the averaged septum thickness of the saline-treated animals within the reversion trial for 15 days were very close to those of the CCl4 + Saline group (all p > 0.05); which indicated that no relevant spontaneous fibrosis resolution took place during the 15 days in which the CCl4 was not injected (Figures 1C and D; table 3).
The therapeutic administration of GHRP6 for 15 days in the first reversion protocol, allowed for 37% of fibrosis clearance (p = 0.0004). It was mainly due to reduction of more than 30% of septae thickness (p = 0.0011). No differences were found in the number of cirrhotic nodules (p = 0.0602) between the groups (Table 3).
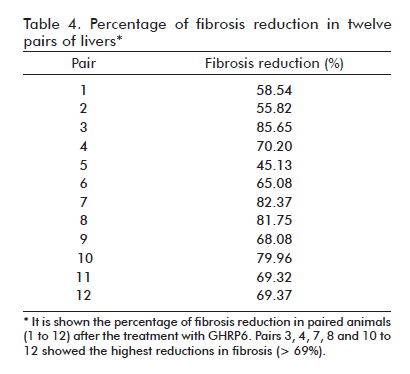
After seven months of CCl4 continuous administration, the CCl4 + Saline-60d group reached the largest percent of fibrosis which was significantly different from the CCl4 + Saline group (p = 0.0139) at the fifth experimental month, indicating a progression in the disease severity due to a substantial increase in septal thickness. As shown in table 3, the therapeutic administration of GHRP6 by 60 days achieved a 75% reduction of the fibrotic area when compared to the Saline-60d group (p < 0.0001), even though the CCl4 injections were not interrupted (Figure 1F). Table 4 shows the reduction in fibrosis from each of the twelve pairs of liver from GHRP6-60d and Saline-60d groups. A reduction higher than 69% is observed in pairs 3, 4, 7, 8, 10, 11, 12 and higher than 80% in pairs 3, 7 and 8. Accordingly, the GHRP6-60d group showed significant reductions in the cirrhotic nodules/mm2 and the septae thickness as compared to the Saline-60d group (both p < 0.0001).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The CCl4 + GHRP6 group exhibited a two-fold increase of hepatic fat percentage with respect to the CCl4 + Saline group (p = 0.0002; table 3). At the seventh month, the GHRP6-60d group showed a 38% increase of fat deposition compared to the Saline-60d group (p = 0.0067). In contrast, in the rats from the GHRP6-15d group, where the CCl4 injections were interrupted, a notorious reduction of fat was detected as compared to the Saline-15d group (p < 0.0001).
Serum biochemistry
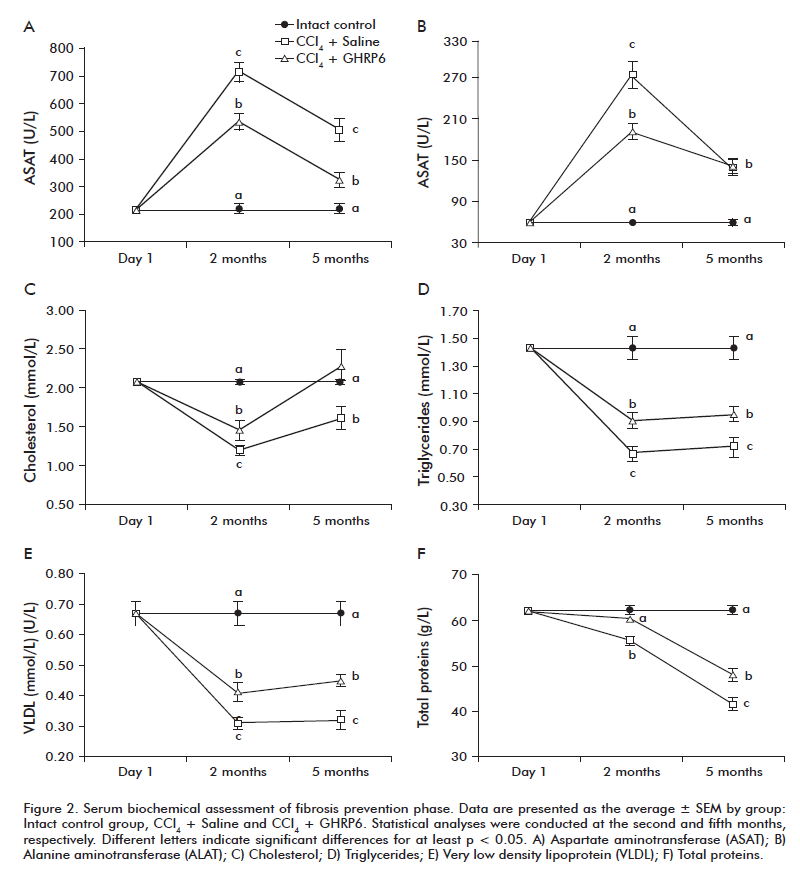
In the fibrosis prevention trial the serum biochemical analysis was done after the second and the fifth month of continuous CCl4 and GHRP6/Saline administrations. At the second month, the CCl4 + Saline group showed the highest ASAT and ALAT levels detected as compared to the Intact control group (both p < 0.001; figures 2A and B). These values dropped by the fifth experimental month although remained significantly superior to the healthy animals (both p < 0.001). A similar biphasic behavior for both transaminases was observed for the CCl4 + GHRP6 group; which resulted significantly lower than those detected for the CCl4 + Saline group at the second month (both p < 0.001). At the fifth month, the ASAT level of the CCl4 + GHRP6 group was also significantly lower than the CCl4 + Saline group (p < 0.001) while similar values for ALAT were observed for both groups. The CCl4-induced liver damage was associated to a reduction of the organ’s biosynthetic function in both groups (Figures 2C to F). However, at the two evaluation time points, the CCl4 + GHRP6 group exhibited a significantly better liver function as compared to the CCl4 + Saline group for all the evaluated parameters (at least p < 0.05).
{kind=link}
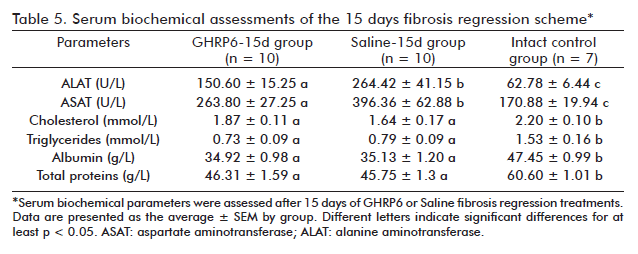
In the fibrosis regression trial, serum chemical parameters in the GHRP6-15d and Saline-15d groups were evaluated once the short therapeutic scheme was completed. As showed in table 5, significant differences for ALAT (p < 0.01) and ASAT (p < 0.05) values were detected between these groups reflecting the GHRP6-induced hepatoprotective effect. Despite this, no significant differences were detected between the GHRP6-15d and the Saline-15d groups in any of the evaluated hepatic synthesis indicators, which remained lower than those of the Intact control group (at least p < 0.05). The serum biochemical parameters from the GHRP6-60d and the Saline-60d groups were also assessed following autopsy (Table 6). No significant differences were detected between these groups for the serum transaminases as for any of the organ’s functional parameters (at least p < 0.05).
{kind=link}
{kind=link}
Hepatic oxidative stress
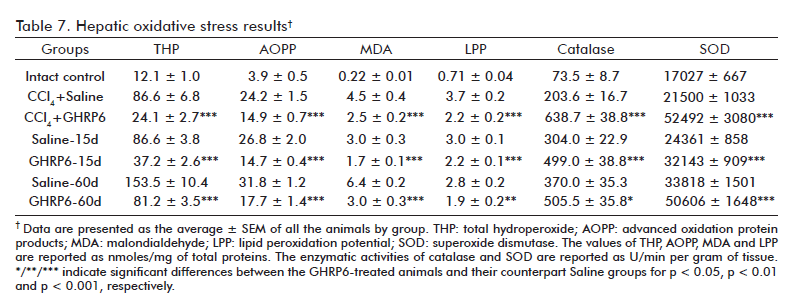
An etiopathogenic ingredient of the CCl4 hepatotoxic mechanism is the oxidative damage to liver cells [24]. It was confirmed by the detected increase of the evaluated oxidative stress markers (total hydroperoxide content, MDA, LPP and AOPP) in all the experimental groups, as compared to the Intact control group (at least p < 0.001; table 7). The GHRP6 intervention significantly attenuated all these oxidative markers as compared with each respective saline group (at least p < 0.01), in the three interventional approaches assessed. Concurrently, the GHRP6-treated rats exhibited a remarkable increase of Catalase and SOD activities as compared to their counterpart saline groups (at least p < 0.05).
{kind=link}
Immunohistochemical results
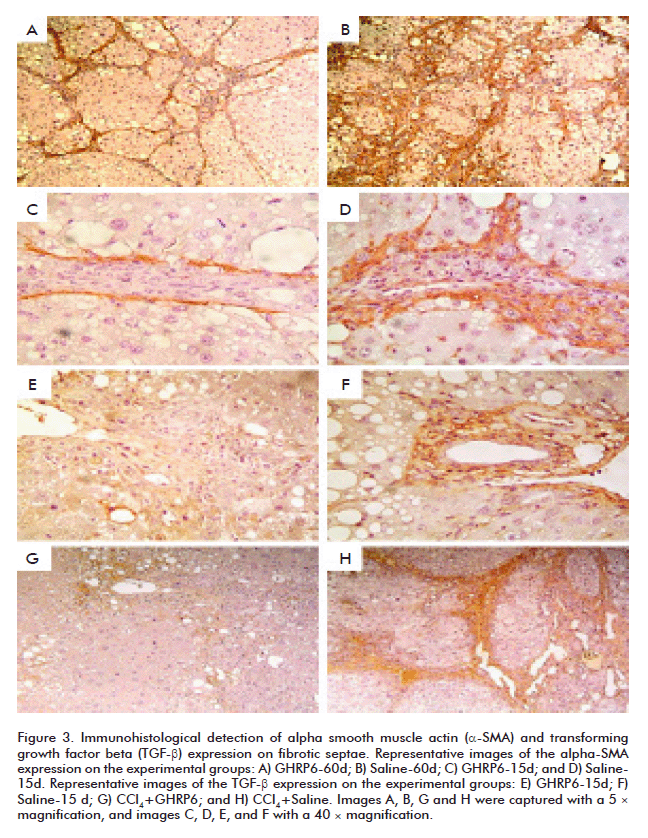
The α-SMA protein is a molecular marker broadly used to detect activated HSC [25, 26]. Although based on qualitative judgment, animals intervened with GHRP6 either under concomitant (data not shown) or therapeutic approaches exhibited far less α-SMA labeling than their saline counterparts (Figures 3A to D). The immuno-detection of the α-SMA antibody appears particularly restricted to the external sides of the fibrotic septae where it has been reported that activated HSC are confined [27]. It is worthy to highlight that an appreciable reduction in the number of labeled cells was a hallmark in the GHRP6-derived samples, including to those fields in which the fibrotic septae appeared similar in terms of thickening and cellular density (Figures 3C and D). Similarly, TGF-β appeared far less expressed in those samples derived from GHRP6-treated rats from both preventive and regression phases (Figures 3E to H). As discussed below, our immunostaining for TGF-β also appeared restricted to the fibrotic septae where it is anchored to the ECM proteins [28].
{kind=link}
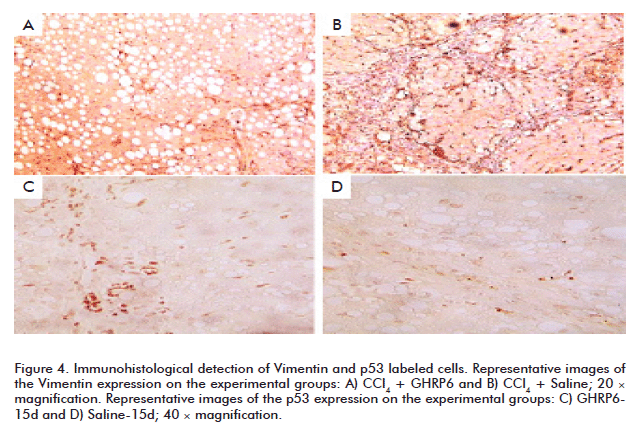
Vimentin is also used as a molecular marker for activated HSC and Kupffer cells [29, 30]. GHRP6-treated animals within the prevention trial showed far less Vimentin positive cells (in the fibrotic septae as within the hepatic parenchyma) than the saline group (Figures 4A and B). This result was also similar for the samples of the fibrosis regression trial (data not shown). As judged by cell morphology and topographic location (core of the fibrotic septae and hepatic parenchyma), lineages immunolabeled with the anti-p53 correspond to recruited round mononuclear cells, Kupffer and HSC. Remarkably, the most intense signal and amount of positive anti-p53 cells were detected in the GHRP6 treated animals; in both prevention (data not shown) and regression trials (Figures 4C and D).
{kind=link}
Although double immunohistochemistry was not conducted, FasL expression specifically matched to Kupffer cells, as suggested by its morphology and location. A larger increase of FasL positively labeled Kupffer cells was detected in the Saline-15d group (14.22 ± 6.17 vs. 2.09 ± 2.12; p < 0.0001). Conversely, the animals of the GHRP6-15d group showed a remarkable increase of Cyclin D1 positively labeled hepatocytes nuclei, as compared to Saline-15d group (8.65 ± 2.36 vs. 2.22 ± 1.59; p < 0.0001), which is likely an indicative of GHRP6-induced parenchymal regeneration [31]. A similar result of FasL and Cyclin D1 expression patterns was obtained in the remaining experimental groups (data not shown).
RT-PCR analysis
The transcriptional profile of fibrosis-committed target genes was studied in the GHRP6-15d and Saline-15d groups (Figure 5). In comparison to intact rats, the saline treated animals showed a significant enhancement on the transcriptional expression of TGF-β and connective tissue growth factor (CTGF; both p < 0.001), two growth factors with well-characterized role in the fibrogenic process [28]. A significantly sharp expression reduction for both fibrogenic cytokines was readily observed in the GHRP6-15d group as compared to the saline counterparts (at least p < 0.05).The CCl4 also induced a significant decrease of the superoxide dismutase manganese enzyme (SODMn) transcriptional expression in the Saline-15d group as compared to the intact control group (p < 0.05; figure 5). However, the SODMn transcriptional expression of the GHRP6-treated rats did not differ from the constitutive levels found in the Intact control group and was significantly superior to the Saline-15d group (p < 0.01). Matrix metalloprotease-13 (MMP13) is the major interstitial collagenase that reduces liver fibrosis by degrading the ECM proteins [32]. A dramatic expression enhancement of MMP13 transcriptional levels appeared with the GHRP6 intervention as compared to the saline group (p < 0.01).
{kind=link}
Microarray and bioinformatic analyses
Microarray experiment was carried out comparing paired samples 4, 7, 8, 11, 12 in Groups GHRP6-60d and Saline-60d with the reference sample from the Intact control group. The 1.5 -fold differentially-expressed rat genes and their homologous human genes (ORT-Human), reported in Homologene or identified by Blastn are shown in table 8. The molecular interaction network that was generated with BisoGenet, using differentially-expressed rat genes as input data, contained only 41 genes and 25 molecular interactions.
{kind=link}
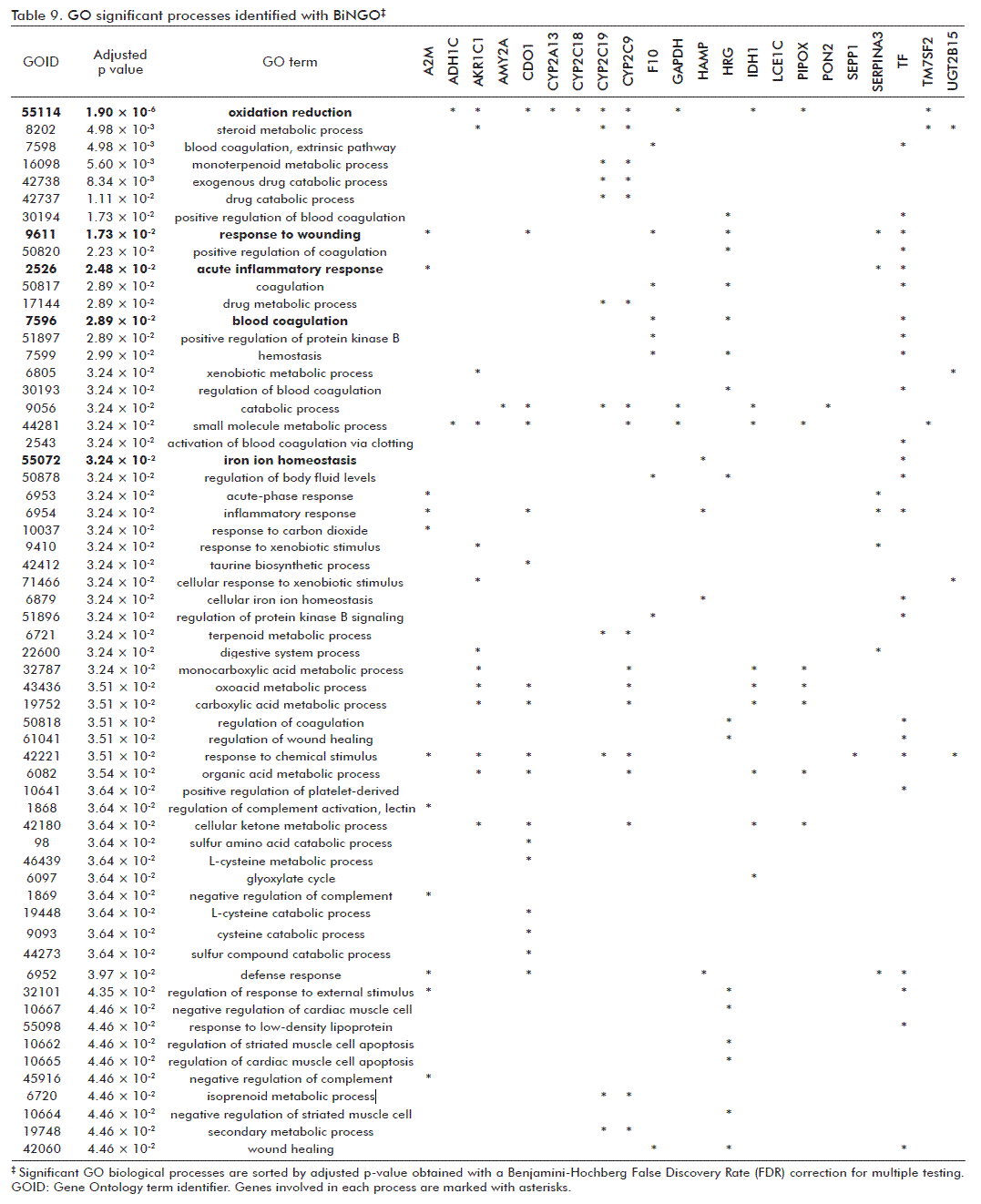
However, a similar network generated from ORT-Human genes included 386 genes and 1883 interactions. After a gene enrichment analysis performed with BiNGO, the most biologically significant processes were underscored as consequence of the GHRP6 intervention. These were: ‘oxidation-reduction’ and ‘response to wounding’ (Table 9). For the former, members of the cytochrome P450 family as CYP2A13, CYP2C18, CYP2C19 and CYP2C9; aldo-ketoreductase (AKR1C1) and UDP glucuronosyltransferase, also committed in drugs and xenobiotic metabolism pathways, were included. Cysteine dioxygenase type I (CDO1), participating in redox process, and the NADP+-dependent isocitrate dehydrogenase 1 (IDH1)/Pipecolic acid oxidase (PIPOX), which participate in peroxisome pathway, were also incorporated within the ‘oxidation-reduction’ process. Besides, Cdo1, alpha-1-inhibitor 3 (A1i3), coagulation factor X (F10), histidine-rich glycoprotein (Hrg), serine (or cysteine) peptidase inhibitor, clade A, members 3N (Serpina3n) and 3M (Serpina3m), transferrin (TF) and hepcidin antimicrobial peptide (HAMP) genes were related to ‘response to wounding’.
{kind=link}
DISCUSSION
CCl4 chronic administration induced an overt cirrhotic disease to otherwise normal rats, which engendered systemic disturbances for the animal homeostasis. Herein, we provide the first evidences suggesting that a classic member of the Bower’s synthetic secretagogue peptides, GHRP6, is not solely endowed with cardioprotective actions but also with anti-fibrotic effect. The evidences derived from these experiments provide the fundamentals to consider that the GHRP6 intervention prevented the progression of a liver fibrogenic process and also triggered its regression. This result substantiates previous findings of our group [11] and others [13] in terms of GHRP6-mediated hepatic tissue protection.
In this study three experimental settings were established. The prophylactic approach attempted to recreate a clinical condition in which a patient is threatened to evolve to fibrosis following a triggering event. Further, the therapeutic intervention trial was split in two clinical schemes, one in which the hepatic challenge was interrupted and an alternative one related to a chronic liver insult. This work seems to be the first preclinical study in which a fibrosis regression effect is examined in homogeneously allocated groups according to a scale of liver gross pathology [15].
The histomorphometric assessment as the ultrasound study concurrently indicated less fibrosis in all the GHRP6 intervened animals. The most remarkable result in terms of fibrosis material reduction was associated to the GHRP6 preventive scheme, where the liver parenchyma appeared spared of nodular organization. Accordingly, the lowest UFI value was calculated for this group. This fact highlights the hepatoprotective effect induced by GHRP6 in order to prevent parenchymal cells downfall and a subsequent fibrogenic response. The short term therapeutic intervention (15 days) accounted for a significant regression of liver fibrosis, mainly expressed in fibrotic septae involution. Importantly, this effect was attained in a scenario where no spontaneous fibrotic resolution took place in the saline group. Parenchymal nodules clearance along with septae thickness reduction was also confirmed in the 60 days GHRP6-therapeutic scheme. It is noteworthy that this effect appeared in a scenario of continuous liver aggression as the CCl4 administration was intentionally maintained.
One of the methodological limitations of this study is the lack of portal venous pressure/flow measurements. Alternatively, the most reliable approach undertaken was to assess portal dilation by ultrasound as described elsewhere [33, 34]. Generally speaking, the incidence of ascites and portal dilation was scarcely registered within the GHRP6 groups, thus suggesting a lesser amount of fibrotic accumulation and a more physiologic haemodynamic performance. We do not rule out however, the hypothetical involvement of the GHRP6 induction of endothelial nitric oxide release as a key factor controlling portal haemodynamic balance [35].
Fat quantification results suggest that a magnification of the fatty liver phenotype is associated to the co-administration of CCl4 and GHRP6 and not to the GHRP6 intervention alone. Long term clinical interventions with the cognate GHRP2 have proved to be safe in dwarf children [36]. In line with this, our long-term systemic administration toxicity studies in healthy rats proved that GHRP6 does not harm the hepatic tissue (Cosme-Díaz K; unpublished data). The Cyclin D1 expression profile incites to speculate that GHRP6 stimulates hepatocytes mitosis. Whether this mitogenic response is directly triggered by GHRP6 itself and/or through the GH/Insulin-like growth factor 1 axis remains to be examined [37]; moreover, regeneration of the hepatic mass could hypothetically explain the increase of fat storage in concomitantly CCl4 + GHRP6-treated rats. It could also explain the noticeable fat storage reduction in those animals solely exposed to GHRP6 without a correlate increase in fat serum markers.
In general terms, ALAT and ASAT serum levels exhibited a biphasic behavior. The acute damage phase, histologically expressed as hepatic steatosis and non-parenchymal cells reactivity (data not shown), correlated with the highest transaminases levels. From this point onward, however, fibrosis severity increase appeared associated to a drastic transaminases drop-down, irrespective to the medication, which has been previously reported [38, 39]. The fact that ALAT levels from the GHRP6-preventively treated group remained similar to the saline counterpart at the fifth month could speculatively be associated to the increase of fat storage and/or to hepatocytes regeneration. These two factors could be also related to the fact that in the regression phase, transaminases were only reduced within the GHRP6-short treatment scheme where CCl4 injections were interrupted. The liver synthesis function remained preserved with the concomitant GHRP6 scheme, as indicative of its hepatoprotective potential and/or to a hepatocytes’ regeneration. However, the liver biosynthetic function did not improve with any of the GHRP6 therapeutic schemes. The CCl4-induced irreversible inhibition of macromolecules exocytosis mechanism in the hepatocytes may explain this drawback [40, 41]. It is reasonable to deem that a more prolonged treatment period, associated to a liver-free insult, would be translated into a superior GHRP6 therapeutic impact in terms of hepatic synthesis function.
Aside from the limitations of this work to fully elucidate the GHRP6 anti-fibrogenic mechanism, a major contribution in this context is the finding that GHRP6 induces an enhancement of SOD activity, which appears mediated via the transcriptional activation of at least the manganese-dependent isoform. In line with this observation, the bioinformatics analysis of the microarray experiment results indicated that GHRP6 enhances the action of a series of genes encompassed in the redox homeostasis as Cdo1. In addition, two other enzymes whose expression appeared upregulated by GHRP6 (PIPOX and IDH1) play instrumental roles in peroxisomal processes as free radicals detoxification. Furthermore, increases of CYP2A13, CYP2C18, CYP2C19, CYP2C9, AKR1C1 and UDP glucuronosyltransferase support the idea of an active detoxification process, contributing to revert fibrogenesis [42]. This has extended previous findings in which GHRP6 proved to ameliorate radicalary cytotoxicity and to amplify antioxidant defenses [11]. As reactive oxygen species are key mediators of the fibrogenic process by directly activating HSC transdifferentiation [28], its neutralization and/or pool reduction has been considered an appropriate strategy to mitigate the fibrogenic responses [43-46].
The inflammatory response associated to the hepatocytes’ injury amplifies the generation of reactive oxygen species, the recruitment of inflammatory cells, as the local release of profibrogenic cytokines [47]. The fact that in the GHRP6 short term therapeutic intervention, Kupffer cells expressed p53 in concomitance to an obvious suppression of FasL expression, may translate as an ongoing process of Kupffer cells arrest or apoptotic induction. It would represent a possible disruption of hepatocytes’ demise and the ensued activation cascade [48]. It is known that Kupffer cells promote hepatocytes apoptosis via FasL pathway [49, 50]. Overall, these observations indicate that GHRP6 contributed to quench the liver inflammatory response as has been previously described [11, 13]. In vitro models are in progress to delineate the actual potential of GHRP6 to prevent HSC activation.
The notion that GHRP6 intervention reduced local inflammation and the consequent HSC activation-perpetuation may be supported by the consistent reduction of α-SMA and Vimentin expression, as markers of an activation phenotype. In line with this, GHRP6-treated animals exhibited far less expression of the two major fibrogenic growth factors TGF-β and CTGF which may anticipate a decrease in ECM protein synthesis and accumulation [28]. Furthermore, GHRP6 increased MMP13 transcriptional expression which may be obviously linked to fibrosis reduction due to collagen degradation [32]. It obviously would be complemented by a functional enzymatic assay.
Growth hormone secretagogues regulate the transcriptional activation of the peroxisome proliferator-activated receptor gamma (PPARγ) through the concerted interaction with the two types of receptors identified: CD36 and the ghrelin GHS-R1a. PPARγ can also induce CD36 expression thus establishing a mutually positive regulatory loop [51]. PPARγ agonistic stimulation markedly inhibited HSC proliferation, induced cells apoptosis and significantly suppressed TGF-β1-induced CTGF expression [52]; which is in correspondence to our immunohistology and microarray data, suggesting an abortion of the HSC activation program. Likewise, Milam and co-workers recently confirmed the PPARγ agonist ligands inhibit the ability of TGF-β to promote myofibroblast trans-differentiation as the ensuing collagen secretion and fibrotic induration in a murine model of lung fibrosis [53]. The above demonstrations render hypothetical mechanistic involvement for the GHRP6-mediated PPARγ activation via CD36 occupation. This may represent a not redundant pathway for novel anti-fibrotic agents.
In summary, the main contribution of this work is the unprecedented demonstration that GHRP6 reduces the fibrotic induration of a parenchymal organ in a renowned experimental model. The mechanism whereby GHRP6 reduces fibroplasias seems to be multifactorial involving a broad number of gene products concerned to detoxification pathways, redox homeostasis, and response to injury. Distally, it appears to nurture hepatocytes and deactivate stroma-associated cells. These preliminary findings justify further studies on the role of the GHS for the control of fibrotic diseases.
ACKNOWLEDGEMENTS
Authors are indebted to Dr. Bienvenido Gra, Head of the Pathology Department from the Institute of Gastroenterology, Havana, Cuba, for his valuable assistance while examining the slides.
REFERENCES
1. Lefton HB, Rosa A, Cohen M. Diagnosis and epidemiology of cirrhosis. Med Clin North Am. 2009;93(4):787-99.
2. Hammel P, Couvelard A, O’Toole D, Ratouis A, Sauvanet A, Flejou JF, et al. Regression of liver fibrosis after biliary drainage in patients with chronic pancreatitis and stenosis of the common bile duct. N Engl J Med. 2001;344(6):418-23.
3. Arima M, Terao H, Kashima K, Arita T, Nasu M, Nishizono A. Regression of liver fibrosis in cases of chronic liver disease type C: quantitative evaluation by using computed image analysis. Intern Med. 2004;43(10):902-10.
4. Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD, et al. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004; 126(7):1795-808.
5. Bourliere M, Kahloun A, Gascou-Tessonnier G. Analogs and fibrosis regression in hepatitis B. Gastroenterol Clin Biol. 2009;33(10-11):923-9.
6. Friedman SL. Liver fibrosis -- from bench to bedside. J Hepatol. 2003;38 Suppl 1:S38-53.
7. Gines P, Cardenas A, Arroyo V, Rodes J. Management of cirrhosis and ascites. N Engl J Med. 2004;350(16):1646-54.
8. Bowers CY, Momany FA, Reynolds GA, Hong A. On the in vitro and in vivo activity of a new synthetic hexapeptide that acts on the pituitary to specifically release growth hormone. Endocrinology. 1984; 114(5):1537-45.
9. Shen YT, Lynch JJ, Hargreaves RJ, Gould RJ. A growth hormone secretagogue prevents ischemic-induced mortality independently of the growth hormone pathway in dogs with chronic dilated cardiomyopathy. J Pharmacol Exp Ther. 2003; 306(2):815-20.
10. Paneda C, Arroba AI, Frago LM, Holm AM, Romer J, Argente J, et al. Growth hormone-releasing peptide-6 inhibits cerebellar cell death in aged rats. Neuroreport. 2003;14(12):1633-5.
11. Cibrian D, Ajamieh H, Berlanga J, Leon OS, Alba JS, Kim MJ, et al. Use of growth-hormone-releasing peptide-6 (GHRP-6) for the prevention of multiple organ failure. Clin Sci (Lond). 2006;110(5):563-73.
12. Berlanga J, Cibrian D, Guevara L, Dominguez H, Alba JS, Seralena A, et al. Growth-hormone-releasing peptide 6 (GHRP6) prevents oxidant cytotoxicity and reduces myocardial necrosis in a model of acute myocardial infarction. Clin Sci (Lond). 2007;112(4):241-50.
13. Granado M, Martin AI, Lopez-Menduina M, Lopez-Calderon A, Villanua MA. GH-releasing peptide-2 administration prevents liver inflammatory response in endotoxemia. Am J Physiol Endocrinol Metab. 2008;294(1):E131-41.
14. Abe W, Ikejima K, Lang T, Okumura K, Enomoto N, Kitamura T, et al. Low molecular weight heparin prevents hepatic fibrogenesis caused by carbon tetrachloride in the rat. J Hepatol. 2007;46(2):286-94.
15. Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The morphology of cirrhosis: definition, nomenclature, and classification. Bull World Health Organ. 1977;55(4):521-40.
16. Witko-Sarsat V, Gausson V, Nguyen AT, Touam M, Drueke T, Santangelo F, et al. AOPP-induced activation of human neutrophil and monocyte oxidative metabolism: a potential target for N-acetylcysteine treatment in dialysis patients. Kidney Int. 2003;64(1):82-91.
17. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article 3.
18. Simon R, Korn E, McShane L, Radmacher M, Wright G, Zhao Y. Design and Analysis of DNA Microarray Investigations. New York: Springer-Verlag; 2004.
19. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498-504.
20. Martin A, Ochagavia ME, Rabasa LC, Miranda J, Fernandez-de-Cossio J, Bringas R. BisoGenet: a new tool for gene network building, visualization and analysis. BMC Bioinformatics. 2010;11:91.
21. Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448-9.
22. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25-9.
23. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57(1):289-300.
24. Biasi F, Albano E, Chiarpotto E, Corongiu FP, Pronzato MA, Marinari UM, et al. In vivo and in vitro evidence concerning the role of lipid peroxidation in the mechanism of hepatocyte death due to carbon tetrachloride. Cell Biochem Funct. 1991;9(2):111-8.
25. Carpino G, Morini S, Ginanni Corradini S, Franchitto A, Merli M, Siciliano M, et al. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig Liver Dis. 2005;37(5):349-56.
26. Tomanovic N, Boricic I, Brasanac D. Immunohistochemical analysis of alpha-SMA and GFAP expression in liver stellate cells. Vojnosanit Pregl. 2006;63(6):553-7.
27. Ramm GA, Nair VG, Bridle KR, Shepherd RW, Crawford DH. Contribution of hepatic parenchymal and nonparenchymal cells to hepatic fibrogenesis in biliary atresia. Am J Pathol. 1998;153(2):527-35.
28. Gressner OA, Weiskirchen R, Gressner AM. Evolving concepts of liver fibrogenesis provide new diagnostic and therapeutic options. Comp Hepatol. 2007;6:7.
29. Wu HH, Tao LC, Cramer HM. Vimentin-positive spider-shaped Kupffer cells. A new clue to cytologic diagnosis of primary and metastatic hepatocellular carcinoma by fine-needle aspiration biopsy. Am J Clin Pathol. 1996;106(4):517-21.
30. Geerts A, Eliasson C, Niki T, Wielant A, Vaeyens F, Pekny M. Formation of normal desmin intermediate filaments in mouse hepatic stellate cells requires vimentin. Hepatology. 2001;33(1):177-88.
31. Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003;15(2):158-63.
32. Watanabe T, Niioka M, Hozawa S, Kameyama K, Hayashi T, Arai M, et al. Gene expression of interstitial collagenase in both progressive and recovery phase of rat liver fibrosis induced by carbon tetrachloride. J Hepatol. 2000;33(2):224-35.
33. Liu Y, Li L, Yu Z, Liu Q, Li Z, Wang Y, et al. Correlative study between portal vein pressure and portal hemodynamics in patients with portal hypertension. Zhonghua Gan Zang Bing Za Zhi. 2002;10(2):135-7.
34. Quintanilha LF, Mannheimer EG, Carvalho AB, Paredes BD, Dias JV, Almeida AS, et al. Bone marrow cell transplant does not prevent or reverse murine liver cirrhosis. Cell Transplant. 2008;17(8):943-53.
35. Fiorucci S, Antonelli E, Morelli A. Nitric oxide and portal hypertension: a nitric oxide-releasing derivative of ursodeoxycholic acid that selectively releases nitric oxide in the liver. Dig Liver Dis. 2003;35 Suppl 2:S61-9.
36. Mericq V, Cassorla F, Salazar T, Avila A, Iniguez G, Bowers CY, et al. Effects of eight months treatment with graded doses of a growth hormone (GH)-releasing peptide in GH-deficient children. J Clin Endocrinol Metab. 1998;83(7):2355-60.
37. Desbois-Mouthon C, Wendum D, Cadoret A, Rey C, Leneuve P, Blaise A, et al. Hepatocyte proliferation during liver regeneration is impaired in mice with liver-specific IGF-1R knockout. FASEB J. 2006;20(6):773-5.
38. Poynard T, Munteanu M, Ngo Y, Moussalli J, Lebray P, Thabut D, et al. FibroTest is effective in patients with normal transaminases, when accuracy is standardized on fibrosis stage prevalence. J Viral Hepat. 2008;15(6):472-3.
39. Sebastiani G, Vario A, Guido M, Alberti A. Performance of noninvasive markers for liver fibrosis is reduced in chronic hepatitis C with normal transaminases. J Viral Hepat. 2008;15(3):212-8.
40. Becker E, Messner B, Berndt J. Two mechanisms of CCl4-induced fatty liver: lipid peroxidation or covalent binding studied in cultured rat hepatocytes. Free Radic Res Commun. 1987;3(1-5):299-308.
41. Poli G, Cottalasso D, Pronzato MA, Chiarpotto E, Biasi F, Corongiu FP, et al. Lipid peroxidation and covalent binding in the early functional impairment of liver Golgi apparatus by carbon tetrachloride. Cell Biochem Funct. 1990;8(1):1-10.
42. Takahara Y, Takahashi M, Wagatsuma H, Yokoya F, Zhang QW, Yamaguchi M, et al. Gene expression profiles of hepatic cell-type specific marker genes in progression of liver fibrosis. World J Gastroenterol. 2006;12(40):6473-99.
43. Shen XH, Cheng WF, Li XH, Xie LM, Sun J, Li F, et al. Effect of antioxidant on hepatic stellate cell proliferation and apoptosis during recovery from liver fibrosis. Wei Sheng Yan Jiu. 2005;34(2):194-6.
44. Di Sario A, Candelaresi C, Omenetti A, Benedetti A. Vitamin E in chronic liver diseases and liver fibrosis. Vitam Horm. 2007;76:551-73.
45. El-Demerdash E, Salam OM, El-Batran SA, Abdallah HM, Shaffie NM. Inhibition of the renin-angiotensin system attenuates the development of liver fibrosis and oxidative stress in rats. Clin Exp Pharmacol Physiol. 2008;35(2):159-67.
46. Ogeturk M, Kus I, Pekmez H, Yekeler H, Sahin S, Sarsilmaz M. Inhibition of carbon tetrachloride-mediated apoptosis and oxidative stress by melatonin in experimental liver fibrosis. Toxicol Ind Health. 2008;24(4):201-8.
47. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209-18.
48. Laptenko O, Prives C. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 2006;13(6):951-61.
49. Pianko S, Patella S, Ostapowicz G, Desmond P, Sievert W. Fas-mediated hepatocyte apoptosis is increased by hepatitis C virus infection and alcohol consumption, and may be associated with hepatic fibrosis: mechanisms of liver cell injury in chronic hepatitis C virus infection. J Viral Hepat. 2001;8(6):406-13.
50. Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123(4):1323-30.
51. Demers A, Rodrigue-Way A, Tremblay A. Hexarelin Signaling to PPARgamma in Metabolic Diseases. PPAR Res. 2008;2008:364784.
52. Sun K, Wang Q, Huang XH. PPAR gamma inhibits growth of rat hepatic stellate cells and TGF beta-induced connective tissue growth factor expression. Acta Pharmacol Sin. 2006;27(6):715-23.
53. Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L891-901.
Received in October, 2011.
Accepted for publication in February, 2012.
Jorge Berlanga-Acosta. Department of Tissue Repair and Cytoprotection. Direction of Biomedical Research. Center for Genetic Engineering and Biotechnology, CIGB.