










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.29 no.2 La Habana Apr.-June 2012
RESEARCH
On the isolation of immunostimulatory active acemannan from Aloe barbadensis
Aislamiento de acemannano immunoestimulador activo de Aloe barbadensis
Mabel Alonso1, Yanet Támbara2, Matilde López1, Julio C Aguilar3, Orestes Mayo4, Enrique Prieto5, José Cremata2†, Gerrit Gerwig6, Hans Kamerling6, Eugenio Hardy7
1 Formulation Development Department
2 Physical Chemistry Department
3 Vaccine Department. Center for Genetic Engineering and Biotechnology, CIGB. PO Box 6162, Cubanacán, Playa, CP 10600, Havana, Cuba.
4 National Center for Biopreparations, BioCen. Beltrán Drive 1½, Bejucal 6048, Havana Province, Cuba.
5 Center for Applied Studies on the Development of Nuclear Energy, Havana, Cuba.
6 Bijvoet Center for Biomolecular Research, Department of Bio-Organic Chemistry, Utrecht University.
7 Nanomat Division, Institute for Science and Technology of Materials, Havana University, Cuba.
ABSTRACT
Acemannan from Aloe barbadensis was obtained with four processes: 1) size exclusion chromatography (SEC) using Sepharose CL-4B matrix followed by ethanolic precipitation; 2) SEC, ultrafiltration using hollow-fiber cartridges (30 kDa or 0.1 µm) and ethanolic precipitation; 3) SEC, precipitation with cetyltrimethylammonium bromide (CTAB) and ethanolic precipitation; and 4) direct precipitation with CTAB and ethanolic precipitation. The detergent CTAB was effective to concentrate chromatographic eluates (process 3) and allowed the direct isolation and purification of acemannan from crude ethanolic extracts (process 4), without recurring to SEC. Process 4 also decreases operation time (9 days vs. 15 days in process 3), and costs regarding raw materials. Both processes generate materials devoid of detectable levels of anthraquinones and contaminating DNA, and proteins below 5% of dry weight. This material was essentially made of mannose; 97% obtained in processes 1-3 and 75% in process 4, with a molecular mass ranging from 2000 to 5000 Mr according to G5000 PW SEC. Acemannan in dry form was sterilized at the optimal 10 kGy γ-radiation dose, and retained both its physical-chemical properties and adjuvanticity for HBsAg co-delivered by the nasal route in mice. The mixture of acemannan-1% benzyl alcohol (w/v) does not affect the adjuvanticity. The total carbohydrates content, SEC-HPLC, pH, microbial limit and organoleptic characteristics of the irradiated polysaccharide suspended in phosphate buffer remained stable at -20 ºC for at least 6 months of storage. These results may be useful for designing processes for producing pharmaceutical quality acemannan to be used in vaccine clinical studies.
Keywords: acemannan, cetyltrimethylammonium bromide, Aloe vera, HBsAg, purification.
RESUMEN
Se obtuvo el acemanano de Aloe barbadensis mediante cuatro procesos: 1) cromatografía de exclusión molecular (SEC) en matriz de sefarosa CL-4B, seguida de precipitación etanólica; 2) SEC, ultrafiltración en cartuchos de fibra hueca (30 kDa o 0.1 µm) y precipitación etanólica; 3) SEC, precipitación con bromuro de cetil trimetil amonio (CTAB) y precipitación etanólica; y 4) precipitación directa con CTAB, seguida de precipitación etanólica. El CTAB fue efectivo para concentrar los eluatos cromatográficos (proceso 3) y permitió el aislamiento directo y la purificación del acemanano a partir de extractos etanólicos crudos (proceso 4), sin recurrir a SEC. En el proceso 4 disminuyeron los tiempos de operación del proceso (a 9 de 15 días del proceso 3), y los costos por materias primas. Ambos procesos generaron acemanano libre de niveles detectables de antraquinonas y ADN contaminante, con niveles de proteína inferiores al 5% del peso seco. El preparado estuvo compuesto fundamentalmente de manosa, 97% en los procesos 1 al 3, y 75% en el proceso 4, con masas moleculares en el rango de 2000-5000 Mr, según SEC en G5000 PW. El acemanano en su formulación seca se esterilizó mediante radiación gamma a la dosis óptima de 10 kGy y retuvo sus propiedades físico-químicas y su capacidad adyuvante, demostrada esta última mediante coadministración con el HBsAg por vía nasal en ratones. La mezcla acemanano-benzil alcohol 1% (p/v) no afectó la capacidad adyuvante. El contenido de carbohidratos totales, las propiedades organolépticas y el perfil SEC-HPLC, el pH y el límite microbiano se mantuvieron estables por al menos 6 meses de almacenamiento a -20 °C. Estos resultados pueden ser útiles para el diseño de procesos de producción de acemanano con calidad farmacéutica para estudios clínicos.
Palabras clave: acemanano, bromuro de cetil trimetil amonio, Aloe vera, HBsAg, purificación.
INTRODUCTION
The Aloe barbadensis plant has a long history of acceptance from the remote civilizations until the present time, as a carrier of several curative properties. The major component of the gel or mucilaginous substance contained in the leaves from A. barbadensis is the polysaccharide acemannan. This polysaccharide is a long chain polymer made of acetylated mannose units, which are held together by β(1→4) linkages [1]. Among the potential therapeutic properties of acemannan are: the acceleration of the scaring of wounds and burns [2], the inhibition of cellular proliferation with autonomous character (antitumoral and anticancerous action) [3] and the antiviral action against a variety of viruses (e.g., herpes simplex, newcastle, measles, and HIV) [4]. Owing to both its stimulative and immunomodulatory direct effect on the immune system, the acemannan polymer has been intended for the treatment of cancer, viral diseases, breathing illnesses, as well as for inflammations and infections [5]. In a wide range of conditions where the final stage of resolution requires the immune system response, the acemannan has been proposed as an adjuvant of other medications [6]. Its use has also been recommended in combination with anti-infective, antitumour, anti-inflammatory and antidepressive substances, without showing toxic effects and with a wide synergic spectrum [7].
One of the more recent suggestions for the application of acemannan is to use this polysaccharide as an adjuvant of antigens administered by means of the oral and parenteral routes [8, 9]. In our laboratories, it was designed for the first time a formulation for vaccine administration via the nasopharyngeal route whose main components are the surface antigen from the hepatitis B virus (HBsAg) and the acemannan polymer [10]. This novel formulation shows, in animal models, mucosal responses that are comparable to that of the anti-hepatitis B vaccine administered through the parenteral way. It has been evidenced, also, that the polysaccharide acemannan can act as an adjuvant for the nasal administration of other soluble antigens of different nature [11].
Given the variety of possible uses and applications of this polysaccharide, it remains necessary that isolation/separation studies are carried out to allow designing operationally viable and scalable productive processes for development laboratories to obtain acemannan for clinical trials. Consequently, it was studied here, at bench scale, the application of four processes for the obtainment of acemannan: a) size exclusion chromatography (SEC) using Sepharose CL-4B matrix followed by ethanolic precipitation, b) SEC, ultrafiltration using hollow-fiber cartridges (30 kDa or 0.1 µm) and ethanolic precipitation, c) SEC, precipitation with cetyltrimethylammonium bromide (CTAB) and ethanolic precipitation, and d) direct precipitation with CTAB and ethanolic precipitation; optimal conditions for CTAB-induced acemannan precipitation are described. These proposed processes were analyzed on the basis of several parameters: recovery, operation time for each step, physical-chemical characteristics and purity of the obtained material as well as acemannan-displayed immunopotentiating properties in the model assay of HBsAg antigen administered throughout the nasal route. Also, electromagnetic γ-irradiation for sterility and storage stability testing of the purified acemannan were evaluated.
MATERIALS AND METHODS
Materials
Biological materials
The Hepatitis B surface antigen (HBsAg), in sodium phosphate-buffered saline solution (PBS 1x) at a purity greater than 95%, was supplied by the Center for Genetic Engineering and Biotechnology (CIGB, Havana, Cuba). Leaves from the A. barbadensis Miller plant were collected in the experimental station for medicinal plants Dr. Juan Tomás Roig (Güira de Melena, Artemisa, Cuba). The collected leaves, which were from plants of 3, 6 or 12 years of age, were classified on the basis of their position in the plant. The leaf that was located in close proximity to the floor was named external, while the one more distant from the floor was called internal leaf. Between both types of leaves was the half level leaf or so-called intermediate.
Chemical reagents
They were of analytical or technical grade, acquired from different commercial suppliers (e.g., Merck, Darmstadt, Germany).
Methods
Extraction of the active chemical substance from the A. barbadensis plant
A collection of 100 plant leaves of more than 3 years of age was defined as a batch of leaves. Each batch was washed with abundant water to eliminate earth residues, and then left for 5 days at 4°C in a dark place. Next, the leaves were decontaminated using 3% (w/v) hypochlorite and the tips, borders and bark of the leaves were eliminated. The resulting gel or mucilaginous substance was homogenized in a domestic blender (Hamilton Beach, USA) and then filtered, under gravity, by using 6-7 sterile gauze layers. To the obtained filtrate, ethanol (95%) was added slowly until reaching an ethanol:gel volume relationship of 4:1. This suspension was left over at 4 °C under slow agitation for 24 h, to be then centrifuged at 6700 x g for 20 min; the resultant supernantant was discarded.
The ethanolic precipitate was resuspended at 6 g/L in a sterile solution of 0.2 M NaCl, using a rotor-stator agitator (IKA-Merck, Germany) that operated at 500 rpm for intervals of 2 min. Next, this was centrifuged at 10 000 x g at 4 °C for 5 min. The resulting supernatant was filtered under vacuum through a Whatman 3MM membrane (Whatman, UK). The concentration of total hexoses for this material was determined by an adaptation of the below-described Anthrone method. The obtained material was divided in 200 mL aliquots that were stored at -20 °C until use.
Purification of acemannan by means of SEC and ethanolic precipitation (process 1)
Aliquots with 200 mL of filtered supernatant were defrosted in a bathroom (IKA-Werk, Germany) at 37 ºC and then they were applied in a column packed with 300 mL of Sepharose® CL-4B gel (Pharmacia, Sweden). The running buffer solution was 0.2 M NaCl, pH 7; the flow used was 30 mL/min. The chromatographic separation process was monitored at 206 nm. For each chromatographic run, fractions coming from the first peak (corresponding to acemannan) were collected in volumes from 1 to 1.5 L.
To the collected eluates, 95% (v/v) ethanol was added, at an ethanol:eluate proportion of 4:1 and then the mixture was kept at -20 °C for 6 h. The precipitate obtained was separated by centrifugation at 6700 x g for 20 min. The sediment was washed twice with 70% (v/v) ethanol, and vacuum dried. The dry material was stored at -20 °C in 50 mL Corning tubes until further resuspension.
The resuspension was carried out in phosphate buffer solution (1.25 mg/mL NaH2PO4 · 2H2O and 1.4 mg/mL Na2HPO4 · 2H2O, pH 7.0) by means of agitation with a rotor-stator (IKA, Germany), until the polysaccharide was resuspended totally. Finally, this material was filtered with Minisart filters (Sartorius, Goetingen, Germany) of 5 µm (pore average) size, to eliminate non-resuspended particles, and in this way to get a homogeneous solution.
Purification of acemannan by using SEC, ultrafiltration and ethanolic precipitation (process 2)
A volume of 3.5 L of collected eluates from the SEC run in the Process 1 was processed in a diafiltration system (model DC-2) from Amicon (Millipore, Billerica, MA, USA), equipped with hollow fiber cartridges of 30 kDa pore size operated at an entrance pressure of 0.7-1 bar. When the final volume of the material in the concentrator glass was of 1 L, the polysaccharide was then precipitated with ethanol as described above.
Purification of acemannan by SEC, precipitation with CTAB and ethanolic precipitation (process 3)
To 3.5 L of chromatographic eluates from the Process 1, 0.2 M sodium tetraborate salt (borax) was added until achieving a final concentration of 0.01 M. The solution was maintained under agitation, at 24 ºC for 20 min. Subsequently, 10% (v/v) CTAB was added, until an appropriate concentration for the precipitation was reached. The resulting solution was retained under slow agitation at 4 °C for an hour. The precipitate obtained from the solution was separated by centrifugation at 8000 x g for 20 min. After that, 150 mL of 0.9 M CaCl2 solution was added and the mixture was homogenized with a rotor-stator (IKA, Germany). The final separation of the polysaccharide precipitated was carried out by adding 4 volumes of ethanol, at -20 °C for 6 h; this was collected by centrifugation at 8000 x g for 30 min.
Isolation and purification of acemannan through direct precipitation with CTAB followed by ethanolic precipitation (process 4)
The crude ethanolic material obtained, as described above, was weighed in the range from 3 to 30 g and resuspended in sodium tretaborate solution (0.01-0.1 M). The resulting solution was kept at 24 ºC under slow agitation for 20 min. Afterwards, a 10% CTAB solution was added and the mixture remained at 4 ºC under slow agitation for 1h. The precipitate attained was collected by centrifugation at 8000 x g for 20 min. To the resultant precipitate, 150 mL of 0.9 M CaCl2 was added, and the mixture homogenized with a polytron (IKA, Germany). To the obtained total volume, 4 volumes of 95% ethanol were added, and the mixture was kept at -20 °C for 6 h. The polysaccharide precipitate was separated by centrifugation at 8000 x g for 20 min. Next, two washes with 70% (v/v) ethanol were carried out, and the final material was dried off under vacuum.
Optimization using a response-surface methodology
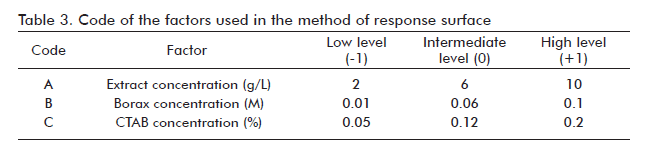
A response-surface experimental design, which utilizes a quadratic model, was created with the use of the Box-Behnken design, and was run using the statistical program Desing-Expert 5 (Stat-Ease, Inc., Minneapolis, MN, USA). Three factors were investigated: the concentration of borax (0.01-0.1 M), the concentration of crude ethanolic material (2-10 g/L) and the concentration of CTAB (0.05-0.2% v/v). Each factor had three levels. The design output was: 17 experimental runs, 11 unique combinations and 6 replicates in the central point (6 g/L, 0.06 M, and 0.12% v/v). The response studied was the precipitation yield, and a polynomial model was defined for the response:

where Y is the response, β0 is a constant coefficient, Xi are the non coded variables, βi are the linear coefficients, βij are the interaction coefficients of second order and βii are the quadratic coefficients.
The data for the response were incorporated in the program Design-Expert 5 for analysis. The ANOVA test of simple factor was used to determine the significance of the results as well as the adjustment of the model. The response surface was built by means of the proposed quadratic model, and an optimum value was found for a maximum of the response.
Sterilization by gamma irradiation of the purified acemannan
This was carried out, in the Center for Application and Development of the Nuclear Energy (CEADEN, Havana, Cuba), by irradiating the product at 25 °C in a PX-gamma 30-type irradiator, with 60Co source at a dose rate of 2.86 kGy/h.
Radiostability of the purified acemannan
Acemannan samples at 1 mg/mL were filled in 2R glass bulbs (hydrolytic quality of class I) from Nuova OMPI, Piombino Dese, Italy, and then they were sealed with chlorobutyl stoppers and flip-off aluminum seals (Helvoet-Pharma, Alken, Belgium). In parallel, other acemannan samples were dried off under vacuum and dispensed in Corning tubes of 15 mL. They were then sealed tightly in nylon bags. Both the bulbs and Corning tubes were divided into 5 groups of 10 units each one. Each group was irradiated at different radiation levels (1, 5, 10, 20 and 30 kGy), after which the samples were analyzed by using: a sterility assay, SEC-HPLC and immunogenicity against HBsAg after antigen delivery through the nasal route.
Stability of irradiated acemannan
The material in dry form, obtained from the Process 1, was resuspended at a concentration range from 0.5 to 1.2 mg/mL in phosphate-buffered solution at pH 7. Next, the suspension was filled in glass bulbs (0.6 mL/bulb); the bulbs were stored under controlled temperature at -20 ºC. Bulbs in representative amounts were removed for analysis at the initial time and at the 20, 30 and 60 days of storage. The samples were analyzed by: microbial limit assay, organoleptic characteristics, pH, determination of total hexoses and molecular homogeneity according to SEC-HPLC.
Analytical methods
Immunopotentiative capacity of acemannan
To evaluate the immunopotentiative capacity of the purified acemannan, 3 immunization schemes that used female Balb/c mice (of around 12 weeks of age and with approximately 20 g of weight) were carried out. The intramuscular route was used as the parenteral route and the nasal via as the mucosal route. The total inoculum volume for the systemic route was of 250 µL and for the intranasal route of 50 µL. For the intranasal delivery, the mice were anesthetized by using an intraperitoneal injection containing 40 µL of ketamine at 10 mg/mL.
Seven groups were used with 10 mice each. To the mice from the groups 1 to 5, two doses with 5 µg of HBsAg and the adjuvant irradiated (0.3 mg/mL acemannan) were given. The irradiation level of the acemannan in each group from 1 to 5 was: 1, 5, 10, 20, and 30 kGy, respectively. The sixtieth group of mice received the same mixture of antigen and adjuvant, with the exception that the acemannan polysaccharide was not irradiated. Another control was based on the inoculation of acemannan-free HBsAg in PBS 1x. All the inoculations were done the days 0 and 14; blood extractions using the retro-orbital via were done on days -2 (preimmune) and 21.
Seven groups, with 8 mice each, were used. The groups 1-4 received two doses of 5 µg HBsAg plus acemannan adjuvant at 0.3 mg/mL. The fiftieth group received the same mixture of antigen-adjuvant, except that the acemannan (control) was not irradiated. In this scheme, the two control groups were based upon: i) HBsAg alone delivered throughout the nasal route in PBS 1X, and ii) HBsAg adjuvanted with alumina (0.5 mg/mL) and delivered subcutaneously. Only the groups 1 and 2 had the acemannan irradiated (at 10 kGy) and the groups 1 and 3 contained 1% (v/v) benzyl alcohol (preserving agent). The days 0 and 14 were set for the inoculations, while days -2 (preimmune) and 21 were programmed for blood extractions using the retro-orbital via.
Five groups of 10 Balb/c mice each were used. The first three groups were inoculated, days 0, 14, and 21, with three doses of 5 µg of HBsAg and acemannan (0.3 mg/mL). The acemannan used in the groups was: group 1, acemannan from the process 1; group 2, acemannan from the process 3; group 3, acemannan from the process 4; group 4, acemannan from the process 4, after being irradiated. The group 5 was inoculated with HBsAg in PBS 1X. Blood extractions by retro-orbital punction were done at days -2 (preimmune), 26 and 54. After extraction, the blood was incubated first at 37 °C for 30 min and soon after at 4 °C for 30 min. Next, this was centrifuged at 3000 x g for 10 min; the serum was extracted and then stored at -20 °C until use. The immunogenicity results were evaluated by ELISA, as described below.
ELISA anti-HBsAg
The levels of specific immunoglobulins against the HBsAg were determined by ELISA. Plates (MaxiSorp,Nunc, Belgium) of 96 wells were coated with HBsAg at a final concentration of 10 µg/mL in 50 mM carbonate-bicarbonate buffer solution, pH 9.6, at 37 °C for 3 h or 4 °C overnight. After washing three times with PBS/Tween-20 (0.05% v/v), the plates were incubated at 37 °C for an hour, or overnight at 4 °C, with 2% (w/v) blockade solution (skim milk in PBS). The plates were washed again and incubated with the samples at 37 °C for 2 h. Polyclonal serums that recognize the HBsAg were used as positive controls, while preimmune serums were used as negative controls. Also, serums from mice inoculated with the adjuvants used in each scheme (placebos) were included. All the serums had replicas and adequate dilutions in a solution of 1% (v/v) PBS/Tween-20 with 1% (w/v) skim milk. The plates were washed several times and then incubated with antibodies (dilution 1:3000) specific against the fraction of mouse immunoglobulin conjugated to peroxidase (Amersham, UK), at 37 °C for 1 h. After the last washing, the plates were incubated with 0.1 M citrate buffer solution, pH 5, containing 0.015% (v/v) H2O2 and 0.1% (w/v) o-phenylenediamine. Fifteen minutes later, the reaction was stopped using 50 µL of 2.5 N H2SO4 and the optical density at 492 nm was measured with a conventional plate reader. Two-fold the average value of the placebos-generated optical density was used as the criterion for seroconversion.
SEC-HPLC
The high-resolution liquid chromatography system (HPLC) used included a 2250 pump (LKB-Pharmacia,Uppsala, Sweden) and a refraction index-based detec-tor (Knauer, Berlin, Germany). The acemannan samples (1 mL) were applied into a SEC column G5000 PW from TosoHaas (Stuttgart, Germany). PBS 1x containing 0.25 M NaCl was used as the running buffer solution; the system was operated at a working flow of 0.2 mL/min. Acquisition and data processing were carried out using a home-made software (BioCrom, version 2.3). The processing report included the chromatogram shape, retention time for each of the detected peaks, as well as the peak area and its relative percent with respect to the total area from all the detected peaks. This technique was used to control the molecular homogeneity of the acemannan polysaccharide, on the basis of the molecular mass: retention time correlation.
Determination of the concentration of total hexoses
The identification and quantification of the polysaccharide acemannan was carried out with the Anthrone colorimetric method [12]. This is based on the spectrophotometric determination of total carbohydrate concentration, after the hydrolysis and acid degradation of sugars-containing carbohydrates, leading to furfural formation, 5-hydroxymethylfurfural and other derivative products formed in the reaction (in a strongly acid medium) with the Anthrone reagent. Glucose at 2 mg/mL was used as a reference pattern; the glucose calibration curve used included concentrations at 20, 40, 60, 80 and 100 µg/mL. The concentration of the sample to be analyzed was determined by linear regression; determinations were done in duplicate.
Determination of dry weight
This was determined to the pure polysaccharides at a hexose concentration of 0.3 mg/mL, in a humidity analyzer MA-30 from Sartorius (Göttingen, Germany); the samples (1 mL) were heated at a constant temperature of 110 °C.
Determination of the concentration of proteins
This was done using a variant of the Bradford method [13]. For this, a patron curve with well-known concentrations (from 0.2 to 1.1 mg/mL) of HBsAg was used; the respective dilutions were carried out in a solution of 10 mM Na2HPO4, 1 mM KH2PO4, 0.1 M NaCl, 2 mM KCl, pH 7.2. Reading of the samples (λ = 620 nm) was carried out in an ELISA plate with the use of a SensIdent Scanner (Merck, Darmstadt, Germany).
Anthracene derivatives
This was kindly carried out by the Center for Research and Development of Medications (Havana, Cuba), by means of a colorimetric procedure for the aqueous extract of A. barbadensis. The colorimetric procedure was based on the reaction of Borntrager. In this reaction, the phenolic groups from the anthracene derivatives in an alkaline solution form colored phenolates, after which the absorbance of the samples was read at 525 nm [14].
Determination of nucleic acids
Samples were analyzed by the drop method, which consisted of applying in an agarose plate 3 µL of a patron curve of DNA and 3 µL of the samples to be analyzed. The samples were dried off for 2 min, after which the agarose gel was observed under ultraviolet light. This is a semiquantitative method that allows checking that the level of contaminants is below a threshold value by visual inspection.
Chemical analysis
Determination of the constituent monosaccharides
The identification of the constituent monosaccharides, the molar ratio between them and the total carbohydrates content of acemannan samples was determined by using Gas Chromatography (GC) coupled to a flame ionization-based detector (FID; model ChrompackCP 9008 gas chromatograph), as well as by using a Fisons Instruments GC 8060/MD 800 system (Interscience).
The samples were subjected to methanolysis in 1 M HCl/methanol at 85 °C for 18-24 h. A standard mixture of monosaccharides (Arabinose, Fucose, Xylose, Mannose, Galactose, Glucose, N-acetylgalactosamine, N-acetylglucosamine and N-acetylneuraminic acid) with 50 nmol of internal standard was treated in the same way that the analyzed polysaccharides. After cooling at 24 ºC, the solution was neutralized by adding 10 mg of solid Ag2CO3. To the neutralized solution, two drops of anhydrous acetic acid were added. After blended, the resulting suspension was kept in the darkness, at 24 ºC for 24 h, with the aim of achieving the re-N-acetylation of the mixture of methyl glycosides. Later on, the silver salts were removed by centrifugation and the samples were then trimethylsilylated (hexamethyldisilazane: trimethylchlorosilane: pyridine, 1:1:5). Quantitative analysis was carried out by injecting 2-5 μL of clear solution on an EC-1 column (30 m x 0.32 mm i.d., Alltech) mounted in a ChrompackCP 9008 gas-liquid chromatograph coupled with FID (GLC-FID). The temperature was programmed from 140-240 ºC at 4 ºC/min. Semi-quantitative and confirmative identification of the trimethylsilylated-(NAc) methyl glycosides was carried out by injecting 0.2 μL of the clear solution on an AT-1 column (30 m x 0.25 mm i.d., Alltech) using a Fisons Instruments GC 8060/MD 800 system (Interscience) and a 140-240 ºC (at 4 ºC /min) temperature program (GC-MS).
1H-NMR spectroscopy
Prior to nuclear magnetic resonance (NMR)-spectroscopic analysis, samples were exchanged twice in 99.9 atom % D2O (Cambridge Isotope Laboratories, Inc), lyophilized and then dissolved in 99.96 atom% D2O. NMR spectra were recorded on a Bruker AMX-500 spectrometer (Bijvoet Center, Department of NMR Spectroscopy) at a probe temperature of 300 K. The hydrogen-oxygen-deuterium signal was suppressed by applying a water eliminated transformed fourier pulse sequence in 1D 1H-NMR experiments. When necessary, the remaining hydrogen-oxygen-deuterium signal was eliminated by convolution of low frequency contributions in the free induction decay by a first order phase correction. Chemical shifts were referenced to internal acetone (d 2.225). Spectra were recorded using a spectral width of 4032 Hz for 1H. All NMR data were processed using TRITON software (Bijvoet Center, Department of NMR Spectroscopy) software.
Methylation analysis
Desalted samples were subsequently permethylated (DMSO, NaOH, 20 ºC, MeI), hydrolyzed (2 M Trifluoroacetic acid, 120 ºC, 2 h), reduced (NaBD4, 2 h at 24 ºC) and acetylated (pyridine, Ac2O, 30 min at 120 ºC). The obtained partially methylated alditol acetates were identified by GLC-MS; 0.2 mL of the clear solution were injected into an AT-1 column (30 m x 0.25 mm i.d., Alltech) mounted in the Fisons Instruments GC 8060/MD 800 system (Interscience).
Microbial limit assay
This assay was aimed to determine the total count of aerobic mesophile microorganisms, mushrooms and yeasts, and the presence of Pseudomona aeruginosa and Staphylococcus aureus. For this, the samples were diluted in peptone saline solution, and spilled in plates containing tryptone soy agar, to be incubated at 37 ºC for 72 h. Finally, the forming colony units presented in each plate were counted. Also, the determination and identification of P. aeruginosa, S. aureus, Escherichia coli and Salmonella sp. was carried out.
Determination of organoleptic characteristics
Sampling of 20 bulbs was carried out at random. The product was observed in a revision cabinet against white and black backgrounds, while illuminating with 20 W fluorescent lamps. The organoleptic characteristics (e.g., color, absence of precipitate) were carefully monitored to know if any change happened in the samples stored under the conditions established for each study.
pH determination
This determination was carried out according to the requirements of the USP 29, 2006 [15]. The measurement was carried out twice and the final value from both measurements was average.
Statistical processing
The statistical analysis of the results was carried out using the Fisher’s F (for the determination of variance homogeneity) and Student’s t (for the comparison of means) tests. In both cases, that significant differences existed was considered when p was smaller than 0.05.
RESULTS AND DISCUSSION
Acemannan purification by means of SEC and ethanolic precipitation (process 1)
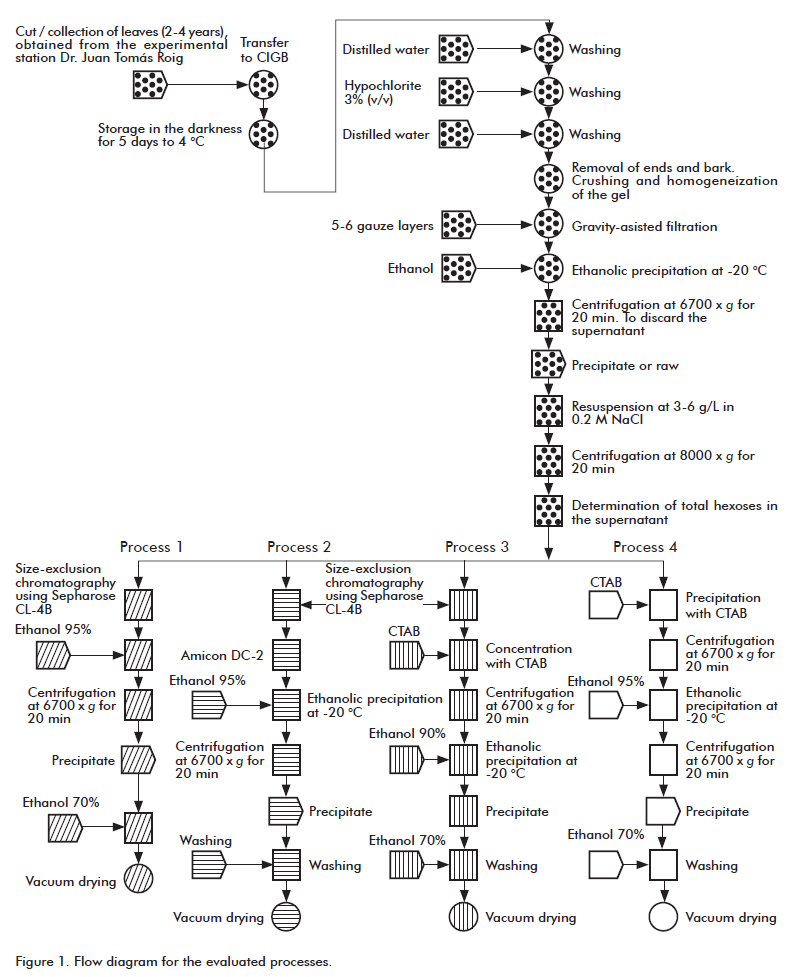
Figure 1 illustrates a flow diagram representing the studied isolation/purification processes for the polysaccharide acemannan (processes 1-4). In general, these processes began with a treatment of the leaves from A. barbadensis and continued with extraction of the active chemical substance by ethanolic precipitation [16]. The resulting material, which was of gray color and fibrous appearance, was resuspended and conserved at -20 °C until further processing.
The purification process 1 is based on the use of SEC, with Sepharose® CL-4B gel, that is appropriate for the expected high molecular mass of the polysaccharide. The collected eluates are precipitated with 95% (v/v) ethanol, and the acemannan was separated in sediment form by centrifugation. Figure 2 shows a typical SEC-based profile; peak 1 belongs to acemannan, whereas peak 2 corresponds to the total elution volume. Because the polysaccharide absorbs poorly at 206 nm, the size (e.g., area, height) of peak 1 does not correlate with the actual total mass of acemannan. The molecular mass of acemannan seems to be between 2000 and 5000 Mr, because its retention time was smaller than that of blue dextrane, a compound with a molecular mass of 2000 Mr (data not shown).
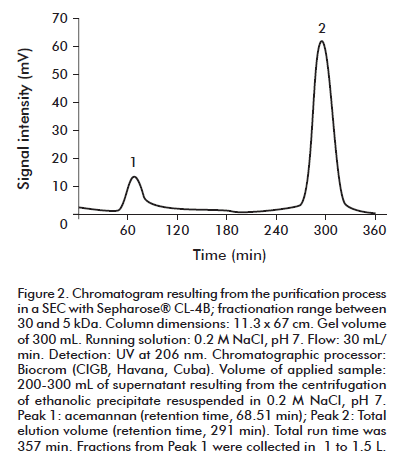
Table 1 summarizes some available average data (operation time and recovery) for the obtainment of an acemannan batch using process 1. Ten chromatographic runs, which correspond to the obtainment of approximately 150 mg of material, were used to constitute an acemannan batch.

Although this process worked well for previous preclinical studies [10, 11], we envisaged that this might be problematic to be scaled-up with the objective of obtaining batches for later stability testing and clinical trials. The chromatographic run of concentrated juice solutions, as starting material from A. barbadensis, is not possible owing to their high viscosity. In fact, two effects associated to the use of very concentrated A. barbadensis juices are: the compression of the chromatographic gel and the unacceptable decrease of the operation flows. Largely diluting the starting material to be chromatographed might solve this problem, but this may imply the processing of bigger material volumes during the ethanolic precipitation step, since an eluate:ethanol relationship of 1:4 is used. These material volumes would become even bigger during the centrifugation stage, significantly lengthening the operation times. All this would rebound adversely in the speed, efficiency and cost of acemannan obtainment if the process were operated at either pilot or production scales.
Consequently, in an attempt to solve these potential problems, it was decided first to evaluate, at a small scale, the introduction of two concentration methods: tangential ultrafiltration and precipitation with the cationic detergent cetyltrimethylammonium bromide. The purpose was to find a procedure that allows concentrating the eluates coming from the SEC step, thus avoiding the processing of big volumes during the ulterior ethanolic and precipitation steps.
Concentration of SEC-eluted acemannan by ultrafiltration (process 2)
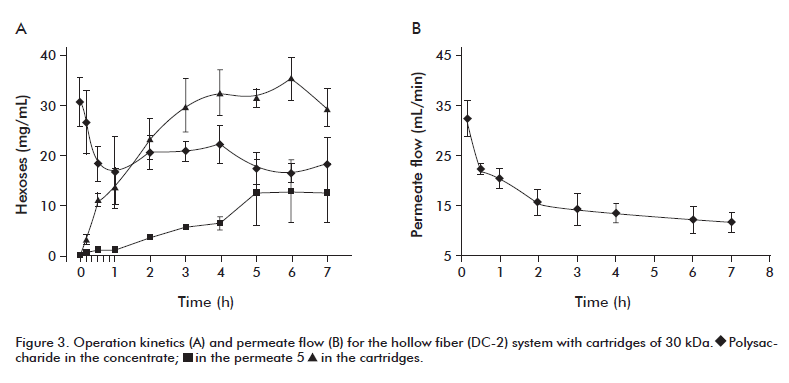
An Amicon ultrafiltration system (DC-2) was evaluated, which was assembled with hollow fiber cartridges of 30 kDa or 0.1 µm pore size. A volume of 3.5 L of acemannan eluate, which resulted from the blending of 3 chromatographic runs, was used as the starting material. The kinetic behavior of the concentration system was evaluated by determining the acemannan quantities escaped in the permeate volume, lost in the cartridges and recovered in the concentration volume. The permeate flow was measured at defined time intervals during the operation. The acemannan yield was determined on the basis of the concentration of total hexoses, at both the beginning and the end of the process.
Figure 3 shows the kinetic behavior of the system during 7 h of operation. With the cartridges of 30 kDa, the yield obtained in the concentrate was about 29%, with 20% losses in the permeate volume. These losses were not associated to the physical integrity of the cartridges, since a control ultrafiltration process with 3.5 L of blue-dextrane (2000 Mr) solution prepared at a concentration similar to that of the chromatographic eluates (0.01 mg/mL), generated a transparent colorless permeate volume, even when the concentrate volume reached 500 mL. Neither the presence of hexoses was detected, either for the 30 kDa or 0.1 µm cartridges. Consequently, the low yields in the glass concentrator were most likely due to the retention of the polysaccharide in the cartridges, with about 49% of mass loss.
This polysaccharide retention in the cartridges could be associated to ‘polarization effects’ due to the different speeds of solute transport, which produces a concentration gradient in the vicinity of the membrane, and an increase of the resistance for interaction of the solute with the membrane causing incrustations [17, 18]. As a consequence, a decrease of the solvent flow per area unit takes place (Figure 3B) and a considerable increase of the operation time and therefore a decrease of the system operability.
With the cartridges of 0.1 µm the acemannan recovery was around 33% after 7 h of operation, and the quantity of polysaccharide retained in the cartridges diminished considerably (Table 2, column 3). However, this happened to expense of considerable losses in the permeate (Table 2). This could be related with the own linear structure of the acemannan that could favor its escape through the cartridges.

From these results, it is possible to conclude that the SEC-eluted acemannan polysaccharide can be concentrated using ultrafiltration systems based on hollow fiber cartridges of polysulfones. Nevertheless, under the evaluated experimental conditions the use of cartridges with a cut off size of molecular weight smaller or similar to 30 kDa is not viable. The use of the cartridges with 0.1 µm pore diameter is not advisable either, in our opinion.
Concentration of SEC-eluted acemannan by precipitation with CTAB (process 3)
CTAB is a cationic detergent, with chemical formula C19H42NBr, which possesses bactericidal properties. In multiple researches, its use has been described for the isolation of capsular or negatively charged polysaccharides derived from different sources [19, 20] for the isolation of mannans [21, 22], as well as for the analytical or preparative isolation of DNA [23]. The polysaccharides in contact with CTAB form an insoluble complex that can be separated by centrifugation.
Here, we thought of the possibility of using CTAB as a precipitating agent for acemannan. Nevertheless, this polysaccharide is constituted by mannose units that confer acemannan the property of being a neutral polysaccharide. For this reason, the acemannan would not be able to interact with the cationic CTAB. This situation would be reversed if negative charges were conferred to the mannose units that form the polysaccharide, and so the acemannan acquired the capacity to form insoluble complexes with CTAB. Guided by this idea, the agent used was borax, with chemical formula Na2B4O7. The function of this chemical reagent would be the formation of a complex for interaction of the borax with the OH- groups in position cis from mannose, thus conferring negative charge to the mannose groups of acemannan.
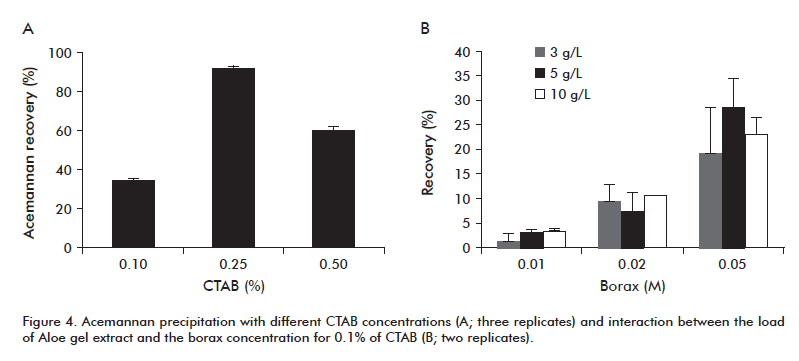
In a first experiment, a concentrated solution of 0.25 M borax was added to 1 L of SEC-eluted acemannan, to a final concentration of 0.01 M, and 10% CTAB (w/v) until getting a concentration of 0.1% (w/v). The recovery of acemannan obtained under these conditions was relatively low (23%), which could be associated to a deficit of appropriate CTAB proportions for the formation of precipitates in the chromatographic eluates. Therefore, different CTAB concentrations were evaluated (0.1, 0.25 and 0.5% w/v), at a constant borax concentration of 0.01 M. After acemannan precipitation, the complex CTAB-borate was separated using a solution of 0.9 M CaCl2. Figure 4A shows that with a CTAB concentration of 0.25% (w/v) the biggest precipitation yield was obtained, which reached a value of approximately 90.3% in terms of total hexoses.
Regardless of the concentration of CTAB used, the recovery of acemannan in the posterior ethanolic precipitation step was constant (data not shown). It should also be noted that the complex CTAB-borate may be disrupted by precipitation with ethanol. This is associated to the high solubility of the detergent in polar solvents [24]. This precipitation-based concentration method for SEC-eluted acemannan would decrease the operation volumes, thus facilitating operational conditions in planned pilot or larger processes.
Replacement of SEC by CTAB-induced precipitation (process 4)
Keeping in mind the potentialities of CTAB and borax in selectively forming complexes with mannose units and the previous results, it was considered the possibility of carrying out the isolation and direct purification of the acemannan polysaccharide in a single precipitation step, starting from the A. barbadensis extract and substituting the SEC step. In this way, we sought to decrease the long operation times of this step and simplify the production process in terms of decreasing the number of steps and unitary cost. The experiments related with this idea are shown here.
The starting material used in the first experiments was the raw gel material from Aloe (15 or 30 g/L), 0.1% CTAB and 0.01 M of borax salt. Acemannan yields (%) were very low in 3 processes of starting concentrations of 30 g/L (11.3 ± 0.43%) and 15 g/L (13.7 ± 3.25%). When adding the borax in the mixture solution an increase was observed in the consistency of the gel, with the form of a lattice. This provoked diffusional difficulties of the reactant substances and could disable the formation of a precipitate when adding the CTAB solution. Given this, small-scale experiments were carried out in which the concentration of starting Aloe gel material (3, 5, 10 g/L) and borax (0.01, 0.02 and 0.05 M) was varied, at a constant (0.1% w/v) CTAB concentration. As shown in Figure 4B, at the concentration of 5 g/L raw material and 0.05 M borax the best recovery results were obtained.
In Figure 5, the process of isolation of the acemannan with CTAB is analyzed using SEC-HPLC. After precipitation with CTAB (Figure 5B), a decrease of the fraction (peak 2) of contaminants was observed. The precipitation with ethanol (Figure 5C), diminished much more the presence of the contaminants in peak 2, increasing the molecular homogeneity about 50%.
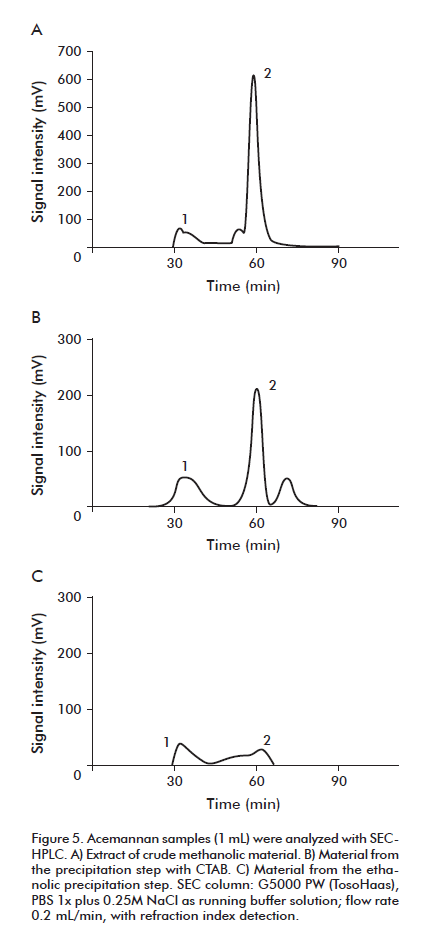
These results suggest that the precipitation procedure with CTAB can be an alternative to the SEC step, to simplify the production process and diminish the costs in terms of gel and chromatographic equipment, as well as the operation time.
Optimization of the conditions for CTAB-induced acemannan precipitation
The variables used for finding the optimum yield of acemannan precipitate were: the borax concentration, raw material load and CTAB concentration. The optimization work was assisted by both the Box-Behnken de sign and a response surface methodology. The latter is a mathematical technique that is used to obtain a designand optimization equation between the measured parameter (response) and a defined quantity of variables (factors) that affect the response. The selected design (Box-Behnken) is quadratic, independent and does not include a fractional or factorial design. For this design, the treatment of the combinations lays in both the midpoints of edges of the process space and at the center. This design is rotatable, requires three levels for each factor and does not have axial points; therefore, all the points fall inside the area for operation. Also, the design allows the work with few points and each factor only requires three levels, instead of the five required for the central composition design. Experimentally for the same number of factors, this design is more convenient and less expensive, in terms of the number of runs, than the central composition design.
Analysis of response-surface regression
The selected parameter of more important response was the precipitation yield, determined by the relationship between the quantity of total hexoses at the end and the beginning of the precipitation process. In table 3, the factors used in the response-surface study are shown, as well as the code that represents each factor. The selected design proposed 17 experimental runs, of them 6 replicas in the central point and 11 in the rest of the points. Table 4 shows the experimental conditions for each run and the yield obtained in the precipitation.
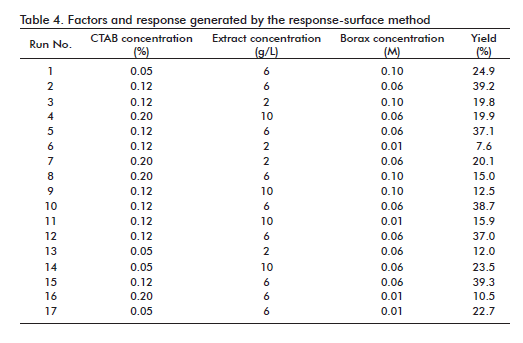
Notice that the response range was from 39.3% in the run 15 (maximum) to 7.6% in the run 6 (minimum). The dependent and independent variables were related by means of a polynomial equation obtained with the Design Expertthe experimental conditions for each run and the yield obtained in the precipitation. Notice that the response range was from 39.3% in the run 15 (maximum) to 7.6% in the run 6 (minimum). The dependent and independent variables were related by means of a polynomial equation obtained with the Design Expertthe experimental conditions for each run and the yield obtained in the precipitation. Notice that the response range was from 39.3% in the run 15 (maximum) to 7.6% in the run 6 (minimum). The dependent and independent variables were related by means of a polynomial equation obtained with the Design Expert ® statistical program (Stat-Ease, Inc. (Minneapolis, USA)):
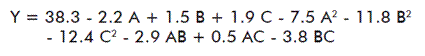
This proposed quadratic model was adjusted to the experimental data, since (p > F) < 0.05, implying that the quadratic term is significant. The adjustment occurred for an appropriate regression value (r = 0.9446). Nonetheless, the test of adjustment that compared the “residual error” and “the pure error” for the points of the design that were replied was significant (data not shown). This indicated that the model should be used with caution when predictions should be carried out.
The next step was to carry out the analysis of surface regression for each factor, according to the use of the code of units. This was based on both the values calculated for the coefficients and the p values (notice that p represents the null hypothesis probability). It was considered that a factor affected the response for a 5% significance level when the coefficient differs significantly from zero and the p value was less than 0.05. The analysis revealed that the precipitation yield was affected more significantly by the quadratic term expressed in the interactions of the CTAB concentrations (p = 0.0057), concentrations of raw material (p = 0.0004) and Borax concentrations (p = 0.0003). From these results, the elimination of the terms with probability values greater than 0.1 could be considered. Under this situation, only the quadratic terms and the interaction BC (borax and CTAB concentrations; 0.0873) were smaller than 0.1. Another important aspect was the low values obtained for the residuals (prediction error), which favors the prediction by the model (data not shown).
Validation of the model
The most important diagnosis for the validation of the model, as well as the confirmation that the suppositions carried out in the variance analysis are appropriate is the analysis of the residuals. For this, two tests are fundamental: the probability adjustment for the residuals and the behavior of the adjustment for the studentized residuals against the predictive response. To sum up, using these tests it was observed (data not shown) that the probability of the residuals is adjustable to a straight line, which indicated that the error is normally distributed, a necessary condition for the variance analysis. In addition, the error terms were independent of the magnitude of the data, indicating that the residuals should be possibly distributed around the line zero with little difference in the width of the treatments.
Analysis of the optimization
With the viewpoint that the proposed model has more than two factors, the factor concentration of CTAB kept constant in the three-dimensional response-surface plot from figure 6. The presence of an optimum and a much defined curvature can be observed, which is characteristic of the quadratic terms from the model. In total, three plots can be produced, if each factor is kept constant. The criterion followed for the optimization was to obtain a maximum of yield for the objective function as response variable. From the proposed model, we found an optimization solution for the maximum yield (0.11% CTAB, 6.30 g/L extract concentration, 0.06 M Borax), accounting for a 38.53% of acemannan recovery and 0.976 desirability.
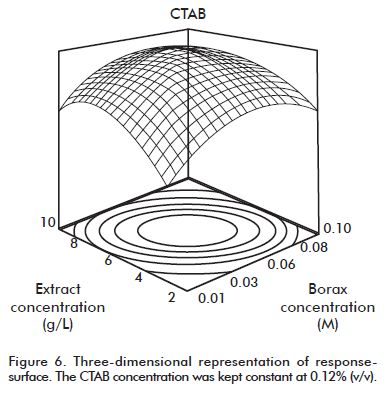
Characterization of the purified acemannan
The product obtained was characterized, in terms of the elimination of major contaminants, and the preservation of both physical-chemical characteristics and immunostimulatory properties of the acemannan.
Elimination of major contaminants
In the process of juice extraction from A. barbadensis, any fibrous material (e.g., cellulose) contained in the juice is also precipitated by ethanol. This happens during an early stage of the precipitation, where the fibrous material, being less dense than the polysaccharide of interest, flocculates in the solvent surface and is eliminated easily [16]. Also, the A. barbadensis juice possesses large quantities of anthracene derivatives (e.g., anthraquinones), organic acids, oligosaccharides, monosaccharides and inorganic salts that can be eliminated in the extraction process, due to the high solubility of these compounds in ethanol [16].
Elimination of anthraquinones and protein contaminants
For this study, polysaccharides derived from batches coming from leaves of plants of 4 to 6 years of age were selected, either for the process 1 [11] or for process 4 based on precipitation with CTAB. Also, leaves from plants of 3, 6 and 12 years of age were collected and classified by their position in the plant, as shown in table 5. The polysaccharide coming from these leaves was obtained by means of the process 1.
The anthraquinones determination was carried out with a validated technique that detects a minimum concentration of 0.9 ppm anthracene derivatives. Regardless of the age and zone of the leaves, all the analyzed samples were below this concentration; consequently, the presence of this contaminant was also below 9·10-5 % of the total dry weight. The data referred to the concentration of this substance in the crude ethanolic materials show that concentrations equivalent to 0.042% (respect to the dry mass of the substance) were present. This indicated that by means of the purification process 4 the anthracene derivatives diminished approximately 500 times. Thus, the acemannan preparations were liberated from most of the contaminants of this nature. The same result was observed when the polysaccharide acemannan coming from different parts of the leaf (internal, in-between and external) was isolated and purified with the process 1.
The results of protein concentrations per dry weight of purified acemannan samples are shown in table 5 and table 6, for the proposed processes. The greater concentration of proteins was found in the process 1 (purification using SEC and ethanolic precipitation). Notice that these procedures for isolation and purification of acemannan were not able to eliminate totally the content of proteinaceous contaminants. Nevertheless, the content of proteins was smaller than 5% per dry weight, according to that described for the use of polysaccharides obtained from different sources [25]. Moreover, it is to be considered that the application of the polysaccharide in this case is for nasopharyngeal use and therefore this quantity of protein should not affect the security of the product.
Elimination of nucleic acid contaminants
All the samples were below 7.5 ng of nucleic acids, representing less than 0.25% of the dry weight. It is inferred, consequently, that all the processes applied for the obtainment of acemannan eliminate nucleic acids efficiently.
Determination of hexoses content
The percent of hexoses with regard to the total weight of purified acemannan oscillated between 31 and 35% (Table 6). This reaffirmed that the major component from the SEC-eluted peak 1 as well as that obtained by the process 4 was of polysaccharidic nature.
Determination of constituent monosaccharides
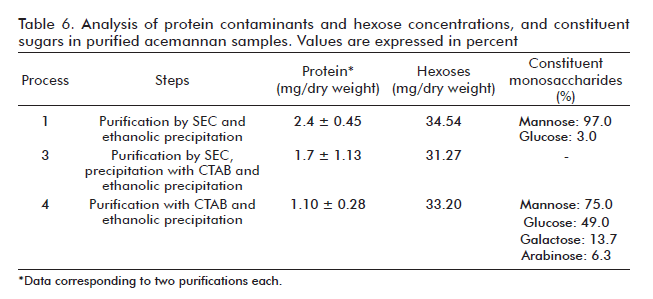
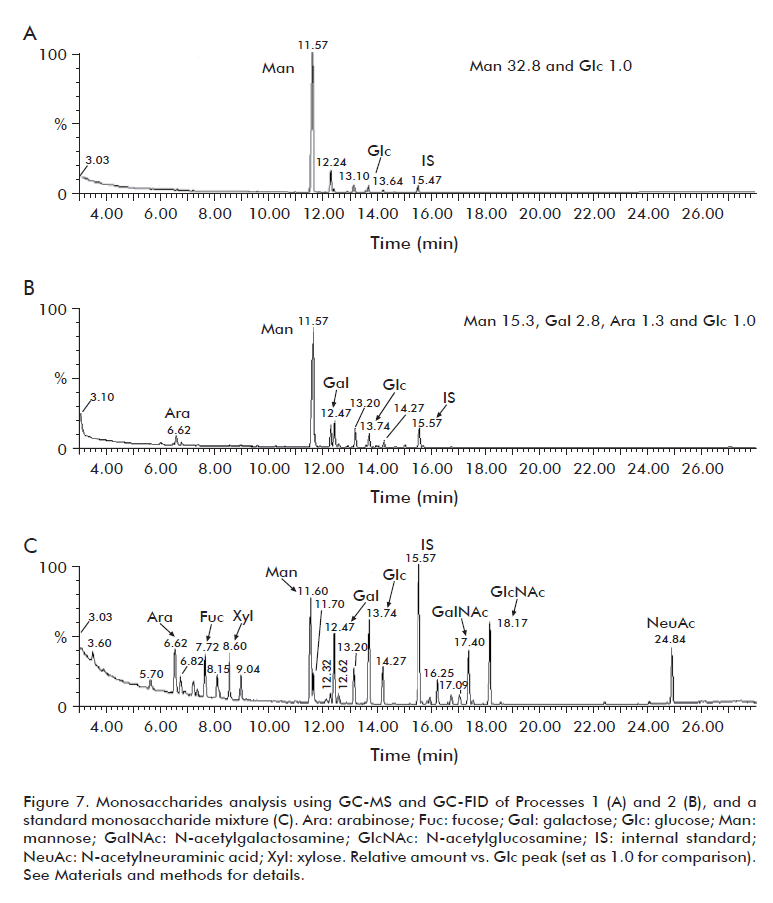
By means of methods based on the use of GC-FID and GC-MS techniques and other chemical procedures (e.g., methanolysis, re-N-acetylation, trimethylsilylation), the composition (monosaccharides) of purified polysaccharide samples was determined for two of the studied processes. As shown in figure 7 and table 6, process 1 separates the acemannan in a more selective way in terms of mannose (97%). Process 4 produced a mannose composition of 75%, N-acetyl mannosamine (ManNAc) was absent and the composition of other produced sugars does not differ from that described in literature [16].
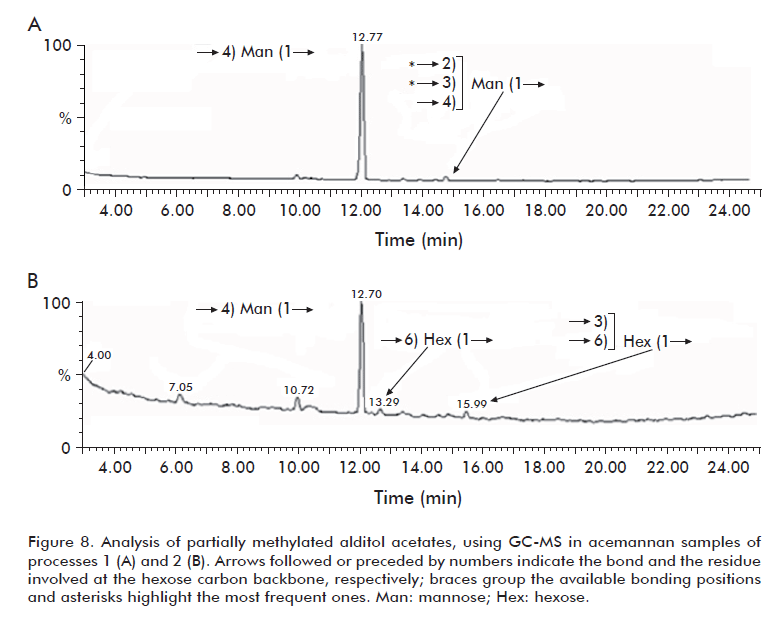
More importantly, the main constituent in both cases was mannose, as expected. Furthermore, some heterogeneous O-acetyl groups but not N-acetyl groups were detected in both polysaccharide preparations, in 1D 500 MHz 1H-NMR spectra (data not shown); to obtain quantitative information (i.e., to know how many O-acetyl groups are present per unit of monosaccharide) is out of the scope of this work. About 95% of this mannose was substituted at position 4, confirming [→4)Man(1→], as determined from the methylation analysis (Figure 8). Despite this showed total coincidence with the backbone previously described [26], improved conditions for the partial chemical hydrolysis of these polysaccharide preparations into oligosaccharides of 4 or 5 units followed by repeated NMR analysis of purified oligosaccharides will be required to get clearer information about the configuration of the acemannan linkages.
Evaluation of γ radiation-based sterilization of the purified acemannan
It was observed that when filtering acemannan polysaccharide samples either through membrane systems or through cartridges of 0.2 µm cut-off size, the pressure of the filtration systems increased drastically. It is most likely that substantial adsorption and appreciable loss of polysaccharide on the membrane from the filters, even using highly diluted solutions occurred. This effect could be related with the own linear structure that presents the polysaccharide, that can compete with the structure of the membranes, impeding its passage through the filters. This phenomenon precluded the sterilization of acemannan by means of the filtration membranes that are otherwise commonly used for the sterilization of protein solutions.
Then, the use of gamma irradiation for sterilizing the purified acemannan was discussed. In fact, this is one of the most effective and frequently used procedures for the sterilization of raw materials and final products. The emission of gamma rays comes from a nuclear source (60Co or 137Ce); this type of electromagnetic radiation is characterized by a high penetration in the material. In the pharmaceutical field, one of the applications of the ionizing radiation has been the final sterilization of biodegradable poly(lactide-co-glycolide) microspheres intended for parenteral use [27]. Thus, a radiostability study for the polysaccharide obtained in solution and in dry form at different irradiation levels (1, 5, 10, 20 and 30 kGy) was carried out.
Organoleptic characteristics of the irradiated polysaccharide
The organoleptic characteristics of the polysaccharide irradiated in solution always changed with every radiation dose used. A dark coloration was observed on the glass vials and the solution turned yellow in color. Also, important changes were observed in the chromatographic profile for SEC-HPLC separated acemannan (data not shown), as well as an appreciable increase of the peak retention time, which was proportional to the increase of the radiation dose. On the other hand, the polysaccharide irradiated in dry form showed a grey appearance very similar to the non-irradiated dry polysaccharide, but with a much more rigid consistency.
Immunopotentiation of the irradiated polysaccharide
The immunopotentiating activity of the acemannan, before and after the treatment with γ radiation, was evaluated by using the nasal route for the delivery of HBsAg. Figure 9A shows that for all the mice inoculated with the polysaccharide irradiated in solution there was seroconversion. However, it was observed a significant decrease (p < 0.05) of the geometric mean of the titles corresponding to the polysaccharides irradiated at different doses, in comparison to the control of non-irradiated polysaccharide (Group 6). This is related directly to the decrease of the immunopotentiating power of the acemannan on the HBsAg (Figure 9B), which is expressed as the relationship of antibody titles induced by the formulations that contain HBsAg and acemannan, in comparison to a control formulation that does not contain acemannan.
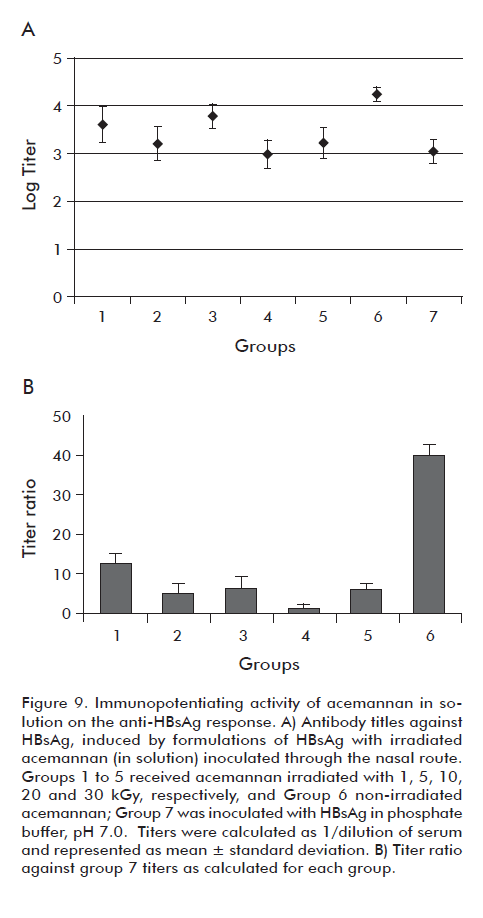
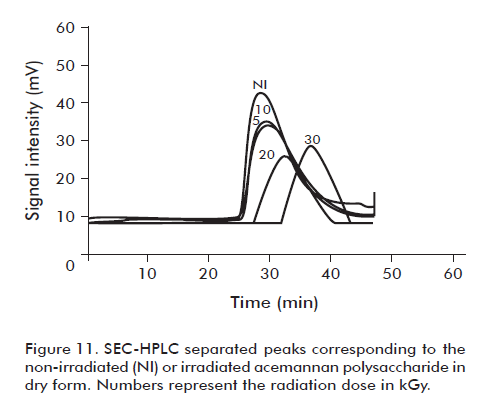
However, the acemannan groups irradiated in dry form did not have significant differences (p < 0.05) among them in terms of their immunopotentiating capacity, for the response of HBsAg with respect to non-irradiated acemannan under the same conditions (Figure 10). On the other hand, it was observed that starting from the 20 kGy, meant a significant increase in the retention time of the acemannan peak (p < 0.05), indicating a decrease in the acemannan size (Figure 11). From these results, the dose of 10 kGy was selected to sterilize acemannan polysaccharide materials in dry form. Under these conditions, the evaluated physical characteristics of acemannan and its immunopotentiating capacity on nasally-delivered HBsAg were not affected. Also, the study demonstrated that at this radiation dose the samples passed the sterility test. Nevertheless, due to the complicated manipulation needed for resuspending the polysaccharide, it was decided in a first work stage to evaluate the acemannan formulations using the microbial limit technique, referred in the Materials and methods section, for microbiological characterization. The parameters followed were: count < 100 c.f.u./mL and total absence of pathogenic bacteria (P. aeruginosa and S. aureus), according to the specifications described in the USP 29 for nasal products.

Immunopotentiation ability of the mixture of irradiated acemannan and benzyl alcohol
With the objective of evaluating the immunopotentiating effect of the acemannan under conditions similar to those that exist in the formulation that would be evaluated in human clinical trials, we projected to evaluate the behavior of the HBsAg immunogenicity response in the presence of both the polysaccharide sterilized by using gamma radiation and 1% (w/v) benzyl alcohol. This preservative agent is commonly used in the preparation of nasal products [28].
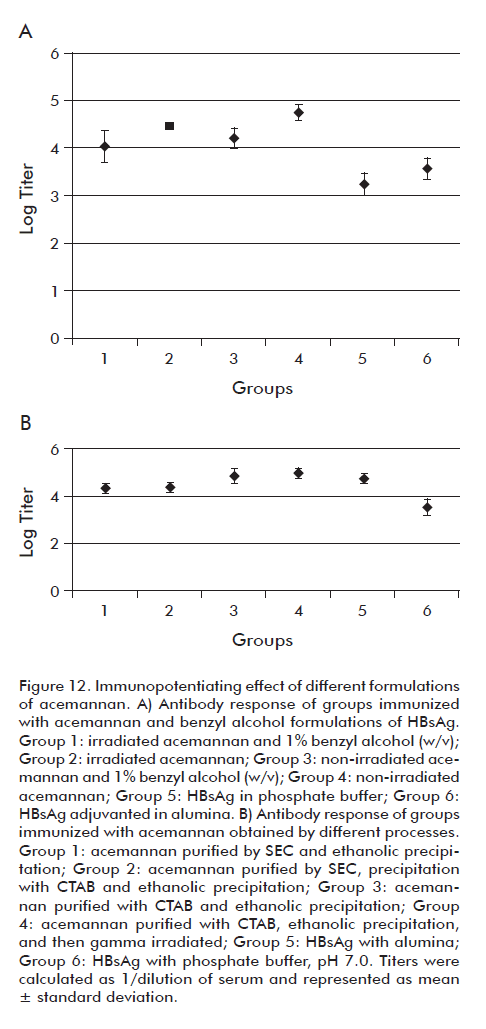
The obtained results are shown in figure 12A. After the first extraction, one week after the second inoculation dose, it was observed that in none of the assayed formulations that contained the irradiated acemannan (groups 1 y 2) there was loss of the polysaccharide-induced immunopotentiating effect. These groups did not present significant differences with the analyzed control groups (groups 3 and 4). It was proven again that the γ-irradiation is a viable method for the sterilization of the polysaccharide, since this does not affect the acemannan immunopotentiating activity on the HBsAg. Also, it was observed that the polysaccharide is not affected, in terms of its activity, by the presence of the preservative (benzyl alcohol at 1% w/v), which supports its use in the vaccine formulation. No significant differences among the groups with preservative (groups 1 and 3) and the groups without preservative (groups 2 and 4) were noticed.
Immunopotentiation of the irradiated polysaccharide obtained by different processes
Aimed to study the immunopotentiating activity of the acemannan purified by the different four processes, we carried out a scheme of nasal immunization in mice, and continued using HBsAg as a model antigen. After the first extraction (one week after the third dose), it was observed that significant differences did not exist in the response of the different acemannan-containing formulations (groups 1, 2 and 3, from Figure 12B). No significant difference was either observed in the immunopotentiating activity of the irradiated acemannan obtained by Process 4 (precipitation with CTAB).
Although the acemannan produced by these processes differed in its monosaccharidic composition (Table 6), the immunopotentiating activity of the result-ing products was very similar in value, which matched with the fact that the main composition of constituent monosaccharides was mannose for all the cases.
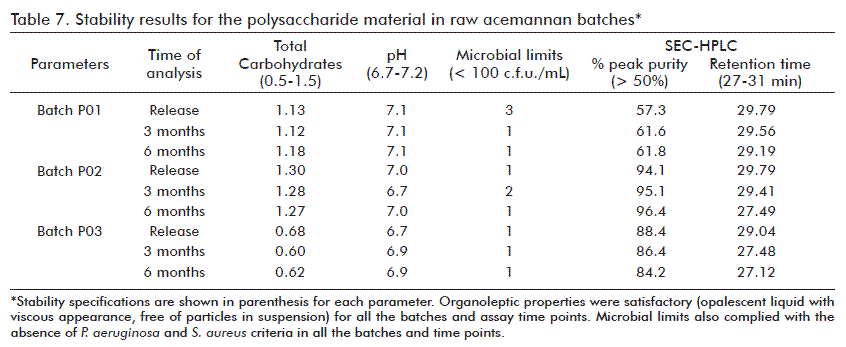
Stability study for the acemannan polysaccharide after irradiation
For this study, three batches produced by using the purification process 1 (SEC and ethanolic precipitation) were used. The batches obtained in dry form were irradiated with the dose of 10 kGy. After being resuspended, the batches were evaluated systematically on release, and after 3 and 6 months of storage at -20 ºC, according to the preliminary specifications of organoleptic characteristics, pH and microbial limit established for the acemannan material (Table 7).
The retention time of the major peak corresponding to the polysaccharide remained constant. There was total absence of P. aeruginosa and S. aureus. From the organoleptic point of view, the solution showed to be an opalescent liquid of viscous appearance, without presence of precipitates or particles in suspension that indicate degradation or decomposition of the product. The pH values remained near neutrality, by which the presence of any degradation product from the polysaccharide that could modify the pH of the solution was discarded. Thus, these parameters remained stable and were within the same limits for the acemannan polysaccharide at the release time.
Further considerations on the processes proposed for the isolation and purification of the acemannan polysaccharide
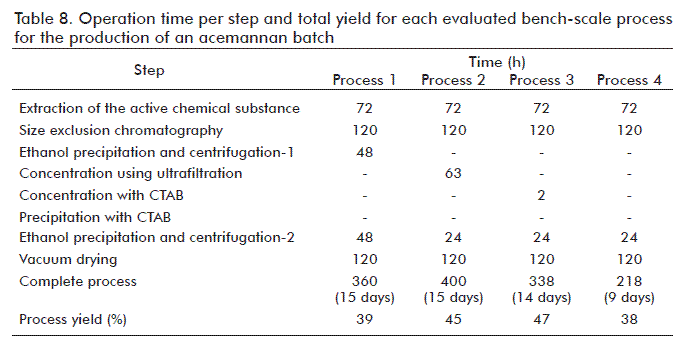
Table 8 shows the operation time and the yield for each stage from all the processes described (Figure 1). In processes 2 and 3, where concentration steps were introduced, an increase of the global yield of the process took place. However, the operation time is still high and similar to that of process 1. In process 4, there was a decrease in the number of steps, and consequently, there was an important simplification of the process complexity for acemannan obtainment, accompanied by a decrease in 40% of the operation time. In this process, the smallest values of protein contaminants were attained. The material obtained in process 4 presented a monosaccharidic composition more heterogeneous than that in processes 1, 2 and 3 that used the SEC separation method. Nonetheless, the immunopotentiating activity of the acemannan did not differ from that of the polysaccharide obtained with the other processes (1-3), indicating that acemannan preparations with a mannose composition similar or superior to 75% conserve its immunopotentiating activity, at least with HBsAg.
CONCLUDING REMARKS
The Amicon ultrafiltration system with cartridges of 30 kDa or 0.1 µm, to concentrate SEC-eluted acemannan polysaccharide (process 2), is not viable due to the high losses that are generated (> 40%). However, the SEC-purified acemannan can be concentrated by means of precipitation with CTAB (0.25% v/v) and borax salt (0.01 M) (process 3). Even more, the acemannan polysaccharide can be isolated and purified directly with CTAB and borax salt, from the crude ethanolic extract of leaves from the A. barbadensis plant (process 4). The experimental data on the isolation and purification of the acemannan with CTAB can be adjusted to a quadratic model, according to the design of Box-Behnken. The optimum precipitation conditions were: 0.12% (v/v) of CTAB, 6 g/L of raw material and 0.06 M of borax salt. The polysaccharide, obtained by means of the four studied processes, does not contain detectable levels of anthraquinones and contaminating DNA. The major component of these materials was mannose and the composition was 97% mannose and 3% glucose for the process 1 (SEC and ethanolic precipitation). Process 4 (precipitation with CTAB) originated 75% of mannose, 49% of glucose, 13.7% of galactose and 6.3% of arabinose. A sterilization method based on the use of the gamma radiation can be used for the acemannan polysaccharide. The polysaccharide irradiated at the dose of 10 kGy does not lose its adjuvant activity for HBsAg, neither in the presence or absence of benzyl alcohol at 1%. The sterilized material showed a long-term stability (at least 6 months at 4 ºC), according to various tests (carbohydrate content, SEC-HPLC, pH, microbial limit, organoleptic characteristics).
REFERENCES
1. Manna S, McAnalley BH. Determination of the position of the O-acetyl group in a beta-(1-->4)-mannan (acemannan) from Aloe barbardensis Miller. Carbohydr Res. 1993;241:317-9.
2. Kaufman T, Newman AR, Wexler MR. Aloe vera and burn wound healing. Plast Reconstr Surg. 1989;83(6):1075-6.
3. Lee JK, Lee MK, Yun YP, Kim Y, Kim JS, Kim YS, et al. Acemannan purified from Aloe vera induces phenotypic and functional maturation of immature dendritic cells. Int Immunopharmacol. 2001;1(7):1275-84.
4. Yates KM, Rosenberg LJ, Harris CK, Bronstad DC, King GK, Biehle GA, et al. Pilot study of the effect of acemannan in cats infected with feline immunodeficiency virus. Vet Immunol Immunopathol. 1992;35(1-2):177-89.
5. Zhang L, Tizard IR. Activation of a mouse macrophage cell line by acemannan: the major carbohydrate fraction from Aloe vera gel. Immunopharmacology. 1996;35(2):119-28.
6. Kemper KJ, Chiou V. Aloe vera: The Longwood Herbal Task Force and The Center for Holistic Pediatric Education and Research; 1999.
7. Kahlon JB, Kemp MC, Yawei N, Carpenter RH, Shannon WM, McAnalley BH. In vitro evaluation of the synergistic antiviral effects of acemannan in combination with azidothymidine and acyclovir. Mol Biother. 1991;3(4):214-23.
8. McAnalley BH, inventor; Carrington Lab Inc., assignee. Use of acemannan. EP 0619117A2. 1989 Aug 3.
9. Nordgrem RM, inventor; Solvay Animal Health, Inc., assignee. Vaccine containing acemannan as an adjuvant. International patent WO/1993/014195. 1993 Jul 22.
10. Petrovsky N, Aguilar JC. Vaccine adjuvants: current state and future trends. Immunol Cell Biol. 2004;82(5):488-96.
11. Aguilar JC, Muzio VL, Leal MJ, Guillén GE, Pentón E, Véliz G, et al., inventors; Centro de Ingeniería Genética y Biotecnología, assignee. Immunopotentiating formulations for vaccinal use. International Patent WO 09839032. 1998 Mar 5.
12. Daniels L, Hanson RS, Phillips JA. Chemical analysis. In: Gerhardt P, Murray RGE, Wood WA, Krieg NR, editors. Methods for General and Molecular Bacteriology. Washington DC: American Society for Microbiology; 1994. p. 518.
13. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-54.
14. Auterhoff H, Schroppel F. Borntrager-reaction with unsymmetrically substituted hydroxyanthraquinones. Arch Pharm Ber Dtsch Pharm Ges. 1969;302(12):937-40.
15. The United States Pharmacopeia. 29th ed. Rockville: United States Pharmacopeial Convention; 2006.
16. MacAnalley BH, inventor; Carrington Lab Inc., assignee. Process for preparation of aloe products. United States patent US4957907. 1989 Jan 25.
17. Martinache L, Henon MP. Concentration and desalting by ultrafiltration. In: Curling JM, editor. Methods of Plasma Protein Fractionation. London: Academic Press; 1980. p. 223-38
18. Genovesi CS. Validation of unique filtration processes. In: Carleton JF, Agalloco JP, eds. Validation of aseptic pharmaceutical processes. New York: Marcel Dekker, 1986:476-506.
19. Inzana TJ, Mathison B. Serotype specificity and immunogenicity of the capsular polymer of Haemophilus pleuropneumoniae serotype 5. Infect Immun.1987;55(7): 1580-7.
20. Kogan G, Jann B, Jann K. Structure of the Escherichia coli 0104 polysaccharide and its identity with the capsular K9 polysaccharide. FEMS Microbiol Lett. 1992; 70(2):135-40.
21. Shibata N, Ichikawa T, Tojo M, Takahashi M, Ito N, Okubo Y, et al. Immunochemical study on the mannans of Candida albicans NIH A-207, NIH B-792, and J-1012 strains prepared by fractional precipitation with cetyltrimethylammonium bromide. Arch Biochem Biophys. 1985;243(2):338-48.
22. Teixeira AZ, Iacomini M, Gorin PA. An unusual glucomannan from Tornabenia intricata. Phytochemistry. 1992;31(10):3467-70.
23. Lander RJ, Winters MA, Meacle FJ, Buckland BC, Lee AL. Fractional precipitation of plasmid DNA from lysate by CTAB. Biotechnol Bioeng. 2002;79(7):776-84.
24. Aspinall GO, editor. The Polysaccharides. Vol 1. New York: Academic Press. 1982.
25. Yu X, Sun Y, Frasch C, Concepcion N, Nahm MH. Pneumococcal capsular polysaccharide preparations may contain non-C-polysaccharide contaminants that are immunogenic. Clin Diagn Lab Immunol. 1999;6(4):519-24.
26. Tai-Nin Chow J, Williamson DA, Yates KM, Goux WJ. Chemical characterization of the immunomodulating polysaccharide of Aloe vera L. Carbohydr Res. 2005;340(6):1131-42.
27. Bushell JA, Claybourn M, Williams HE, Murphy DM. An EPR and ENDOR study of gamma- and beta-radiation sterilization in poly (lactide-co-glycolide) polymers and microspheres. J Control Release. 2005;110(1):49-57
28. Joseph A, Rencher W, inventors; Schering-Plough Healthcare Products, Inc., assignee. Nasal spray compositions. United States patent US5854269. 1998 Dec 29.
Received in December, 2010.
Accepted for publication in March, 2012.
Eugenio Hardy. Nanomat Division, Institute for Science and Technology of Materials, Havana University, Cuba. E-mail: ehardy@imre.oc.uh.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}