










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.31 no.2 La Habana Apr.-June 2014
REVIEW
Dipeptidyl peptidase IV and its implication in cancer
Dipeptidil peptidasa IV y su implicación en el cáncer
Yarini M Arrebola1, 2, Hansel Gómez3, Pedro A Valiente-Flores2, Maria de los A Chávez2, Isel Pascual2
1 Instituto Nacional de Gastroenterología. Calle 25 e/ H e I, Vedado, Plaza de la Revolución, CP 10400, La Habana, Cuba.
2 Centro de Estudios de Proteínas (CEP), Facultad de Biología, Universidad de La Habana, UH. Calle 25 No. 455, Vedado, CP 10400, La Habana, Cuba.
3 Departamento de Química, Universidad Autónoma de Barcelona, 08193 Bellaterra, Barcelona, España.
ABSTRACT
Dipeptidyl peptidase IV (DPP-IV, EC 3.4.14.5), also known as CD26, is a serine aminopeptidase that preferentially cleaves Xaa-Pro or Xaa-Ala dipeptides from the N-terminus of oligopeptides and processes regulatory peptides in vivo, leading to their biological activation or inactivation. The ezyme is a homodimer and each subunit is formed by a αβ-hydrolase domain and a β-propeller domain, involved in the enzymatic activity and its interaction with other proteins. It has an important role in multiple physiological functions, including the regulation of glucose metabolism being one of the current targets for the treatment of type II diabetes mellitus. This enzyme also regulates immune system responses mediated by CD4+ T lymphocytes, and recently has been identified a high/low DPP-IV activity regarding physiological levels, in pathologies like thyroid, ovarian, lung, skin, prostate cancers and central nervous system tumors. For these reasons, this enzyme evolves as a new target of attention for the development of more efficient diagnostics being considering as molecular markers for some pathologies and a target for the development of new therapeutic assessments in cancer. Current research interests are focused on depth in the structure-function relation for this enzyme, as a key point for the development of new therapies in pathologies involving DPP-IV activity or its interaction with other proteins.
Keywords: dipeptidyl peptidase IV, serine peptidases, cancer.
RESUMEN
La dipeptidil peptidasa IV (DPP-IV, EC 3.4.14.5), también conocida como CD26, es una aminopeptidasa de tipo serino con preferencia de corte por la secuencia Xaa-Pro o Xaa-Ala, presente en el extremo amino de los oligopéptidos, que procesa péptidos regulatorios in vivo, y provoca su activación e inactivación. Es un homodímero y cada subunidad consiste en dos dominios: αβ-hidrolasa y propela-β, implicados en su función enzimática y su interacción con otras proteínas. Esta enzima interviene en varios procesos fisiológicos relacionados con el metabolismo de la glucosa, por lo que es uno de los blancos para el tratamiento de la diabetes mellitus tipo 2. Además regula la respuesta inmune mediada por linfocitos CD4+, y recientemente se identificó una alteración de su actividad (elevada o muy baja), en relación con sus niveles fisiológicos normales, en varios tipos de cáncer: de tiroides, ovario, pulmón, piel, próstata, tumores del sistema nervioso central, entre otros. Por tales razones y por considerarse un potencial marcador molecular de varias enfermedades, constituye un foco de atención para el diagnóstico del cáncer y el desarrollo de terapias para combatirlo. Muchos son los estudios encaminados a una mayor comprensión de su relación estructura-función como base para el diseño de tratamientos a aquellas enfermedades en cuyo mecanismo molecular interviene la DPP-IV o interactúa con otras proteínas.
Palabras clave: dipeptidil peptidasa IV, peptidasas serino, cáncer.
INTRODUCTION
Proteases are involved in a myriad of physiological cellular processes, including growth, differentiation, nutrition, protein turnover, migration and diapedesis, fertilization and zygote implantation, programmed cell death, and others. They also mediate
physiopathological events such as: cancer, neurodegenerative, respiratory and cardiovascular disorders, parasitic infestations, and viral and fungal infections. Hence, the proteases systems have to be tightly controlled by effective metabolic mechanisms, with proteases inhibitors as one of the key mechanisms. Inhibitors are widely distributed throughout all the biological kingdoms, and they are responsible for halting inadequate proteolysis and its tuning. Under normal conditions, they guarantee partial proteolysis as a physiological event. Moreover, since proteases are crucial mediators in the replication and infectivity of several pathogens in man, plants and animals, the development of specific and efficacious inhibitors for potential therapeutic application has emerged as an active research field [1-3]. They have been found as effective therapeutic tools in cancer, the human immunodeficiency syndrome (AIDS), inflammation, cardiovascular and respiratory diseases, Alzheimer’s disease, and type 2 diabetes mellitus [1-3].
Particularly, the serin proteases (SP) comprise the best characterized family of proteases due to exhaustive studies conducted in the last 50 years with kinetic, chemical, physical and genetic techniques. A remarkable example is dipeptidyl peptidase IV (DPP-IV, EC 3.4.14.5), also known as complement differentiation protein 26 (CD26), a SP belonging to the prolyl-oligopeptidases with a cell surface expression pattern. It bears a wide anatomic distribution, with its highest specific activity in the kidney [4]. Besides, a soluble isoform is present in several body fluids [5].
DPP-IV selectively removes the aminoterminal dipeptide from peptides having proline or alanine in the second position. Various cytokines, growth factors and some neuropeptides bear this structural motif, what contributes to their respective biological activities and their protection against unspecific proteolysis [4]. Additionally, there are two peptide hormones naturally targeted by DPP-IV as substrates which are determinant in mammalian metabolism: the glucagon-like peptide type 1 (GLP-1) and the glucose dependent insulinotropic peptide (GIP). This makes DPP-IV a new target for therapeutic intervention in type 2 diabetes mellitus.
DPP-IV can also interact with several other proteins, such as adenosine deaminase (ADA), the gp120 protein of the human immunodeficiency virus (HIV), fibronectin, collagen, the chemokine receptor CXCR4 and the CD45 tyrosine phosphatase [6]. This last enzyme also bears several functions aside its enzymatic activity (EA), some related diseases like cancer.
Consequently, DPP-IV has raised a considerable interest in the scientific community: there are a climbing number of publications every year describing its multiple functions, in fields so varied as endocrinology and neuroendocrinology, immunology and oncology [6].
GENERAL PROPERTIES OF DPP-IV
Anatomical distribution, chromosomic location and gene regulation
Few proteases have been described which may be able to escind the post-proline peptide bond, particularly if that residue is located in the second aminoterminal position of the polypeptidic sequence. The posproline aminopeptidase family comprises six proteins of the dipeptidyl peptidase (DP) family: DPP-IV, the fibroblasts activation protein (FAP), DPP-8, DPP-9, the dipeptidyl peptidase-like protein 6 (DPL-1; also known as DPP-6) and the inactive dipeptidyl peptidase 10 (DPL-2; also known as DPP-10) [6, 7].
DPP-IV (EC 3.4.14.5) was initially described as glycilproline naftylamidase, by Hopsu-Havu and Glenner [8] in a commercial preparation of acylase I from rat liver, and further denominated DPP-IV or posproline dipeptidyl peptidase [9]. It was subsequently isolated from various mammalian tissues, in bacteria and plants [10-14]. This aminopeptidase is identical to the CD26 molecule, a surface marker in b and T lymphocytes, and also a protein binding ADA. Moreover, DPP-IV exists as a cell surface protein and is characterized by its ubiquity, being found in humans in epithelial cells in the liver, intestines and kidneys. A soluble form is also found in body fluids, and its expression is regulated in B and T lymphocytes [15]. The highest specific enzymatic activity of this protease is found in the seminal fluid [6, 16, 17] and the kidney [6, 18].
Its human gene is located in the large arm in chromosome 2 (2q24.3), spanning approximately 70 kb and including 16 exons of 45 bp-1.5 kb in length [19], containing domains and transcription factor binding sites for constitutive genes [20]. In spite of the single mRNA identified for DPP-IV [21], a significant heterogeneity has been found in the protein once expressed, possibly caused by postranscriptional modifications [22].
DPP-IV is expressed as a highly glycosylated, type II integral membrane protein [6, 23, 24]. Its natural dimeric and soluble form is present in the seminal fluid, saliva and bile, and derives from the cell surface CD26 molecule, starting from the S39 residue [25, 26]. The release mechanism is unknown, although it has been assumed as being proteolytic [27]. Its serum levels in healthy adults reach approximately 22 nmol/min · mL of p-nitroaniline, equivalent to 7 µg/mL [18].
Molecular structure of DPP-IV
This protein is normally found as a homodimer of 220-290 kDa molecular weight [18, 28, 29], also forming tetramers of around 900 kDa. Each monomer consists of two domains, a αβ-hydrolase (residues 39-51 and 501-766) and a β-propeller domain (residues 59-497) (Figure 1A). There are nine N-glycosilation sites, most of them located in the β-propeller domain, near to dimerization surface. It has been proposed that glycosylation shield the enzyme from extracellular proteolysis [18]. Human and porcine enzymes are similar in size (766 amino acids) with an 88 % homology and share functional properties such as: stability against pH and temperature changes, and susceptibility to peptidases and divalent ions, what makes porcine DPP-IV an adequate surrogate model when the human enzyme is not available due to ethical or economic reasons [30]. Some of the properties of the human and porcine DPP-IV have been recently described for the rat counterpart, indicative of a highly conserved structure-function relationship of this enzyme in mammals [31].
Tridimensional structure
The elucidation of the tridimensional structure of DPP-IV, based on obtaining crystals for structure characterization studies, was fostered by the growing interest in designing inhibitors specific for this enzyme [28, 30, 31].
DPP-IV active site
The catalytic domain of DPP-IV is formed by a β-sheet of 8 strands flanked by 12 α-helixes, a structural motif known as αβ-hydrolase domain [32]. The active site can be accessed through a lateral gap of approximately 15 Å through the cavity where it is located [33]. For this reason, only unfold peptides and partially unfold protein fragment can reach it. Hydrolysis products are released through the tunnel formed by the β-propeller domain (Figure 1B).
The catalytic triad (S630, D708 and H740) located in the interphase between the αβ-hydrolase and β-propeller domains (Figure 1 C). Residue Y547, outside this triad, is also essential for the enzyme’s activity and seems to stabilize the reaction intermediary tetrahedral oxyanion [31]. There are two glutamate residues in the catalytic pocket (E205 and E206) contributing to align the peptidic substrate to the binding site, through salt bridges with the aminoterminus of the peptide to be excised. These residues just make room for two amino acids, what determines the dipeptidyl aminopeptidase nature of the enzyme. Data obtained from mutations of E205 and E206 residues allowed to establish its relevance for the enzyme catalytic activity [34, 35]. Furthermore, its presence is a molecular fingerprint of the DPP-IV family of proteins.
The second aminoterminal residue in the substrate can only be a small sidechain amino acid, such as proline, alanine or glycine, the only ones that could fit in the narrow hydrophobic pocket S1 of DPP-IV formed by residues V711, V656, Y662, Y666, W659 and Y631 [31]. This further determines the substrate specificity of the enzyme.
Homodimerization is a requisite for the catalytic activity of DPP-IV. That process involves the αβ-hydrolase domain [35] and the bulge of the fourth sheet of the β-propeller. A point mutation near the C-terminus of the protein, for example H750 to E, is enough to halt the enzyme dimerization [36].
β-propeller domain of DPP-IV
The β-propeller domains are formed by four to eight β-sheets of 30-50 amino acids each, organized in four antiparalel strands [37]. Those β-sheets are radially displaced from a central tunnel of approximately 30-45 Å, forming a highly simmetric structure. This type of domain was firstly described for the influenza virus neuraminidase [37], which bears six β-sheets. Afterwards, other enzymes were described carrying this domain, such as: the methylamine dehydrogenase [38] and the galactose oxidase [39], both with seven β-sheets, and the methanol dehydrogenase [40] with eight. The number of proteins identified as carrying this domain has considerably grown since 1998, the properties of their supramolecular structures been subsequently described by Murzin [41], Fülöp and Jones [42], Paoli [43], and Jawad and Paoli [44].
β-propellers commonly serve as scaffolds for protein-protein interactions [42, 45] and also mediate in the catalytic activity of the enzymes carrying them [46, 47]. Particularly, some of those enzymes are related to the pathogenesis in some diseases, as in cancer, Alzheimer’s disease, Huntington disease, arthritis, familial hypercholesterolemia, rethinitis pigmentosa, arterial hypertension and also infections [48].
The structure of DPP-IV is unique by having a β-propeller domain of eight β-sheets, compared to the other two leucocyte surface molecules carrying a β-propeller domain, of seven β-sheets each: CD100 [49] and the integrin α chain [50]. Its domain is distinctively disorganized among those described and the eight β-sheets are displaced forming a 30-45 Å in diameter cavity [5]. Since DPP-IV is a type II integral membrane protein, this structural domain is exposed to the extracellular milieu, its structure influencing the interaction of the molecule with other molecules such as ADA, HIV gp120, fibronectin (FN) and collagen [23].
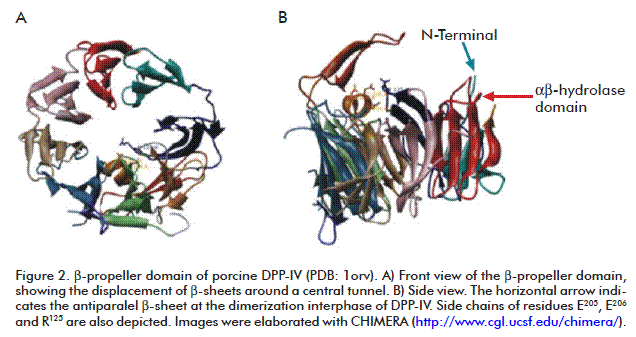
One depicting element in the DPP-IV β-propeller domain is an antiparalel β-sheet which inserts between strands 1 and 2 of the second β-sheet of the enzyme. That antiparalel β-sheet contains the R125 residue, which forms a salt bridge with E205. This last residue locates at the C-terminal turn of the W201-E205 α-helix which intrudes between β-sheets three and four in the β-propeller domain. Another antiparalel β-sheet is located between strands 3 and 4 in the fourth β-sheet, comprising residues D230-N263. This antiparalel β-sheet is essential for the dimerization interphase and is also involved in the substrate binding process [31] (Figure 2).
Residue R125 establishes contacts with both substrates and inhibitors, and is a common target for the design of molecules inhibiting the enzyme activity [18, 28, 31, 32, 51-53]. This residue is highly conserved in DPP-IV structure throughout species, from bacteria to humans. At the same time, the sequence motif of the α-helix bearing the E205 residue (D-W-X-Y-E-E205-E-X) is conserved in the entire DPP-IV gene family [54].
DPP-IV and cancer
The progressive loss of cellular and molecular regulatory mechanisms that occurs during carcinogenesis promotes alterations on key cellular processes, which ultimately determine the raise of malignant phenotypes displaying: autonomous cell growth, irresponsiveness to growth inhibitory signals, cell death evasion, unlimited replicative potential, sustained angiogenesis, tissue invasion and metastasis [55]. Most of these alterations are related to abnormal cell signaling circuits, with overexpressed or constitutively expressed oncogenes, or tumor suppressor genes with null or decreased expression. Particularly, most of those circuits are triggered by molecules secreted by the tumor or its microenvironment. In this context, the auto, para and yuxtacrine regulations determined by growth factors, cytokines, hormones and peptide signals are determinant for the altered abovementioned processes, with the abundance of these ligands depending on the extracellular proteolytic rate which is essential for tumor evolution [56].
It is known that DPP-IV participates in peptide-mediated growth regulation and differentiation and in the regulation of extracellular matrix interactions [6]. The regulation of the DPP-IV-mediated proteolysis could have marked effects on the availability of growth promoting or inhibitory factors in a given microenvironment [6, 56, 57]. Therefore, the loss or lack of DPP-IV expression, and its expression or that of its ligands in the tumor neighboring cells which can be crucial for the progression and metastasis events in several tumor types. The evidences of such events are multifactorial, and their interpretations depend on the properties of the carcinogenesis affected tissues (Table).
DPP-IV expression is decreased in several cancers: melanoma [58], lung [60-62] and prostate [63, 79, 80] cancers, and in serum of oral [81] and colorectal cancers [82, 83]. It has also seen as progressively decreasing during endometrial adenocarcinoma [69]. The opposite effect has been shown in other cancer types, such as: primary lung tumors [59], prostate [63], ovarian carcinoma [70], thyroid carcinoma [64], dermal basal cell carcinoma [71], esophageal adenocarcinoma [72], B-cells chronic leukemia [73, 74] and certain types of T cell cancers (T-cell lymphoblastic lymphoma, anaplastic large cell lymphomas and T-cell acute lymphoblastic leukemia) [6, 75].
It has been further demonstrated the involvement of DPP-IV in the interaction with extracellular matrix components in cancer cells. Its binding to type II plasminogen (Pg 2) on the surface of the 1-LN prostate cancer cell line lead to increased intracellular Ca2+ concentrations, with downstream activation of a transduction pathway ultimately resulting in increased cytosolic pH. That pathway may be triggered by phospholipase C activation which promotes the synthesis of inositol 3,4,5-triphosphate, a well-known inducer of endoplasmic reticulum Ca2+ release [84]. Moreover, it is possible that Pg 2 may regulate pH though its association to the NHE family Na+/H+ exchanger (NH3E) previously bound to DPP-IV. These evidences suggest that the DPP-IV-Pg2 may regulate simultaneously Ca2+, Na+ and H+ concentrations required for tumor proliferation and invasion [84].
The binding of DPP-IV to a subset of extracellular matrix proteins is probably mediated by the β-propeller domain [85]. It was shown the affinity of DPP-IV for type I collagen and FN. So far, the most significant interaction between DPP-IV and FN seems to be that reported during the colonization of the lung by blood-derived cancer cells. Cheng et al. [86] demonstrated that the vascular arrest of metastatic cells in the lung was mediated by the adhesion of DPP-IV to the FN in the surface of cancerous cells. The FN gene is overexpressed in cells able to colonize the lung derived from several cancers in humans, rats and mice. Such metastatic behavior relies on the ability of FN to randomly and dispersedly self-polymerize on the surface of numerous lung cancer cell types [87], and to assemble into long, fibrilar strands. This event occurs by the exposure of FN consensus recognition sequences to the DPP-IV molecules present in the endothelia [86]. It has been demonstrated that interaction depends on SP activity [86]. By the contrary, DPP-IV displays a weak binding activity to plasma soluble FN, suggesting that polymerized FN acquires a conformation different from that of plasma FN [86].
Several findings have confirmed the involvement of DPP-IV and polyFN in lung metastasis: 1) a soluble peptide mimicking the extracellular region of DPP-IV was able to suppress the adhesion of DPP-IV to breast metastatic cells in lung, preventing colonization; 2) the abundance of polyFN in lung metastatic cells, demonstrated in human rat and mice cancers, and also in mice and human melanoma cell lines able to colonize this organ; 3) the polyFN expression in rhabdomyosarcoma clones correlates with lung metastasis [88]. Still remains to be elucidated if the vascular compromise in lung metastasis is solely mediated by the DPP-IV to polyFN interaction or their complexation with other adhesion molecules, such as: proteoglycans, CD44 or heparin sulphate [86].
Binding experiments with a peptide bearing the FN-III repeat 14 sequence (peptide FN-III14) and the native DPP-IV showed that it was able to compete for the binding of polyFN to DPP-IV in the MTF-7 cell line, deriving in profound antimetastatic effects in lung due to 50 % decrease in adhesion and a reduced number of colonies and their size. Such a behavior was similar to that obtained through blocking the DPP-IV extracellular domain by a specific antibody [86]. Another peptide bearing the FN-III14 sequence (22-mer peptide) was described as inducing antimetastasis effects in spleen and liver colonization in T cell lymphoma [89].
DPP-IV in melanoma
In melanoma, DPP-IV is expressed in melanocytes both in vitro and in vivo, but not by the melanoma itself. Its loss of expression seems to occur at a very early stage during melanocyte transformation into melanoma. Wesley et al. [58] demonstrated that DPP-IV-transfected melanoma cells displayed no tumorigenicity or anchorage-independent growth, this last relying on DPP-IV enzyme activity. Additionally, the protein re-expression led to the reacquisition of growth dependency on exogenously provided growth factors [6, 58].
DPP-IV in lung cancer
Lung cancer development relies on the confluence of different growth factors, such as: neuropeptide Y (NPY) and substance P, DPP-IV substrates both. The excision and subsequent inactivation of NPY abbrogate its growth promoting effects [6, 90, 91]. This suggests that the loss of the DPP-IV proteolytic activity would promote growth in certain lung tumor cells, even without confirmation of DPP-IV acting through the regulation of other processes or signaling pathways independent of its enzyme activity, or mediated by other surface molecules as PAF, which stromal abundance correlates with increased tumor cell survival [61].
Although certain lung carcinomas express DPP-IV, that is not the case in large, small-large and small cell carcinomas, with null or marginal expression [60]. In non-small lung cell carcinoma, such lack occurs both at mRNA and protein expression levels (decreasing its activity to less than 40 pM/min/µg of protein) [61], due to frequent losses of chromosome 2q which bears the DPP-IV loci [92-94].
Wesley et al. [61] have proven that the human non-small cell lung cancer cell lines H28, H226, H441, SK-LUC-8, SK-LUC-17, SK-LUC-13, SK-LUC-9 and SW-900 show diminished DPP-IV expression. The restitution of DPP-IV in the line SK-LUC-8, particularly attractive by its undetectable expression of the enzyme, significantly reverted the malign phenotype, independent of the enzyme activity: morphological changes in vitro (long and slightly dendritic cells, adopting cylindric or flat epithelial shape), inhibition of growth in culture (with a lag for the entry in the logarithm phase), inhibition of anchoring-dependent growth (a decreased ability, 50-70 %, to form colonies in soft agar), reduced in vitro migration and cell confluence (probably due to the appearance of density inhibition at a confluence higher than 50 %). This was also related to the increased expression of p21, a drastic cell cycle arrest on G1 and apoptosis induction (possibly by the inactivation of unknown peptides). Simultaneously, there was a high expression of CD44 and PAF, cell surface proteins associated to suppressed growth and metastasis [95, 96]. The subsequent implantation of these cells on athymic mice allowed to corroborate tumor growth as expected, compared to the control which was grafted with tumor cells from the same cell line but untransfected with DPP-IV [61].
Recently, the marginal expression of DPP-IV in A549 (lung adenocarcinoma) and SK-MES-1 (squamous cells carcinoma) cell lines was corroborated; nevertheless, certain variations arose from the history of the carcinogenic process. A549 exhibited an 8-10 times decrease in the overall DPP-IV activity, with a 93 % surface relative activity. This indicates a fast externalization of the recently synthesized protein (probably due to preserved secretory activities inherent to alveolar type II pneumocytes, the original cell type for this cell line).
The whole proteolytic activity of DPP-IV on SK-MES-1 decreased, following a granular intracellular deposition pattern with a 35 % surface relative activity, consistent with the low secretory potential of the cell line. These observations suggest that the distribution of the enzyme during carcinogenesis correlates with alterations that may originate in the intracellular membrane trafficking system [62].
DPP-IV in ovarian cancer
The presence of DPP-IV in ovarian cancer and its involvement in tumor adhesion to the mesothelium was demonstrated by Kikkawa et al. [70]. They showed that the SKOV-3 cells attached more efficiently to mesothelial cells when the DPP-IV expression was restituted. A marked increase in the adhesion to immobilized FN and collagen was also detected.
The mesothelium adhesion effect was shown to be dose-dependent in vitro, compared to soluble FN. That suggests that DPP-IV is a key protein in the tumor cell adhesion to the mesothelium, proliferation and invasion. Ovarian carcinoma and the peritoneal mesothelium where this tumor spreads out, also express DPP-IV, and high amounts of soluble FN and the fibroconnectin of the extracellular matrix are normally found in ascites and the malignant serum. This leads to assume that DPP-IV captures high amounts of FN from these fluids. As a result, carcinoma cells would develop an easy adhesion capacity to either endothelium or mesothelium, once displaying fibroconectin in amounts enough to bind the DPP-IV molecules on these two layers [70].
Nude mice inoculated with SKOV-3 tumor cells reconstituted with DPP-IV showed lower peritoneal dissemination of the tumor cells and longer survival than those receiving the non-reconstituted cell line [97]. Although the causes for such a phenomenon were not completely understood, it would be speculated that the high levels of DPP-IV would promote a tight cell-to-cell adhesion, which may limit the ability of the carcinoma cells to detach from the tumor and spread away from it. Nevertheless, when they detach, the DPP-IV expressed by the tumor and the mesothelium facilitate the invasion into the peritoneum [70].
It could be predicted at least with low probability that the sustained and increased activity of the enzyme could influence indirectly that event, in spite of its innaparent involvement in cell adhesion [70].
DPP-IV in prostate cancer
The benign prostate cancer progresses to a fatal hormone refractory stage through a process considered to be mediated by the overexpression of peptidic growth factors which trigger alternative mitogenic signals [98-102]. Under normal physiological conditions, DPP-IV participates in cell growth regulation and differentiation by regulating those growth factors [103, 104] (e.g., the factor 1-derived stromal chemokine [63]).
Other evidences suggest that prostate cancer metastasis is associated to the loss of DPP-IV (above 50 % in most of the cases) [63, 79] and the increase in the basic fibroblast growth factor activity (bFGF), this last a potent mitogen and pro-angiogenic factor [98-102, 105] expressed as two isoforms, one cytoplasmic of low molecular weight and another one of high molecular weight in the cell nucleus. bFGF transduces through the extracellular signal-regulated mitogen-activated protein kinase (MAPK)-kinase (ERK1/2) pathway, promoting cell cancer progression and migration [106, 107]. Besides, ERK1/2 activation by bFGF during cell migration and angiogenesis increases the production of urokinase-type collagen activator (uPA), an SP catalyzing the conversion of plasminogen in plasmin, and further promoting metastasis through the destruction of the extracellular matrix [108, 109].
It has been proven that DPP-IV restitution in the DU-145 cell line blocks the nuclear translocation of bFGF and the expression of both isoforms, abroggating the stimulation through the MAP-ERK1/2 pathways. This leads to a decrease in uPA mRNA levels, the acquisition of flat and cube-like cell shapes, the loss of contact and anchoring-dependent growth and tumor migration [80].
Although the mechanism by which DPP-IV affects bFGF production remains to be elucidated, it is speculated that in a normal cell, DPP-IV excises its aminoterminal region, causing its confinement within the nucleus. That excision could be the first step for bFGF degradation [80]. Similarly, a direct association may occur with bFGF, that interferes its postranscriptional modification such as methylation of the aminoterminal region, which is required for its nuclear confinement [110-112].
The expression of DPP-IV in DU-145 cells also stimulated transcription of the P27 gene, an inhibitor of cyclin-dependent kinases, halting the cell cycle at the G2-M transition and increasing apoptosis from 24 to 34 % [61].
DPP-IV and thyroid gland: neoplasias, papillary and follicular carcinomas
Thyroid and follicle carcinomas are highly positive to DPP-IV screening, in contrast to benign neoplasias which are markedly negative [64, 67]. Furthermore, DPP-IV expression increases during benign adenoma progression to malignancy, demonstrated by its higher expression in follicle adenoma displaying incomplete capsule invasion compared to that without capsular invasion [65]. The expression of the enzyme is so distinctive at both stages that it is currently considered that DPP-IV levels are the most effective fingerprint to discriminate between follicle carcinoma and follicle adenoma, even more precise that canonical variables such as the patient age, lesion size, its ultrasound image and serum tiroglobulin levels [68].
DPP-IV and keratinocyte tumorization
Experiments in InvEE transgenic mice, bearing keratinocytes with constitutively activated MAP kinase 1 (MEK-1), evidenced the upregulation of DPP-IV in epithelial tumor keratinocytes, particularly notorious at the edge of intercellular contact. Noteworthy, although dermal fibroblasts associated to tissue damage also shown upregulated DPP-IV expression, it was downregulated in the tumor stroma. It was demonstrated that such activity was stimulated by Ca2+-induced intercellular adhesion, and the upregulation of the enzyme in dermal fibroblasts was promoted by addition of interleukine-1α (IL-1α). DPP-IV inhibition reduced tumor growth, and tumor incidence or its delayed appearance in healthy individuals was decreased by blocking IL-1α activity [113].
DPP-IV and neural tissue: gliomas, meningiomas and neuroblastomas
Healthy human brain tissues display a DPP-IV activity mostly considered as mediated by DPP8 and DPP9. Otherwise, in gliomas, the most significant tumor type in the central nervous system with more than 50 % of the tumors, this enzyme activity has been correlated with decreased DPP8 and DPP9 activity, and a spike in DPP-IV and PAF expression, in parenchymal and vascularized areas. At the same time, a high expression of CXCR4, receptor for the stroma-derived factor (SDF-1α), was also found [76]. SDF-1α is one of the endogenous substrates of DPP-IV, and its active form the main chemokine mediating glioma survival [114]. Once excised, SDF-1α loses its chemotactic properties and could even act as CXCR4 antagonist [115]. The marked CXCR4 upregulation seen in glioma would seem to compensate DPP-IV overexpression, suggesting a potential cross-regulation between both molecules [76].
It was found that WHO type I and atypical type II meningiomas express DPP-IV at very low levels, in detriment of increased DPP8 and DPP9 activities. The differential DPP-IV expression in meningiomas and gliomas could reside in their embryonic origin and, paradoxically, could be one of the underlying causes for the lowest aggressiveness of mningiomas compared to gliomas. In fact, meningiomas express normal levels of CXCR4, in agreement with the decrease DPP-IV expression [77]. Thus, the putative ‘compensatory effect’ does not seem to be activated due to insufficient DPP-IV activity, a mechanism that is present in glioma through the SDF-1α activation pathway.
Several human neuroblastoma cell lines show notoriously low DPP-IV expression. The re-expression of the enzyme in vitro leads to the loss of the malignant phenotype: neuron-like or flat epithelium morphology, inhibition of proliferation, caspase-activated apoptosis, decreased Akt phosphorilation and MMP9 activity (known effectors of the SDF-1α-CXCR4 activation pathway) and decreased cell migration. The low proliferation seems to be caused by the induction of differentiation as evidenced by morphological changes. The loss of migration activity may be related to the simultaneous contribution of morphology recovery and the under-regulation of MMP9, this last a proangiogenic factor displaying gelatinase activity on the extracellular matrix [78].
DPP-IV, GLP-1 and cancer
The presence of the GLP-1 factor was demonstrated in the human pancreatic carcinoma Hs-7766T and human pancreatic duct adenocarcinoma CAPAN-1, CFPAC-1 and PL45 cell lines. Nevertheless, there are differences in the stimulation, mediated either through ERK1/2 activation or AMPc induction [114]. Since transduction pathways triggered by ERK1/2 and, to a lower extent those of AMPc, are relevant in events such as mitosis, meiosis and carcinogenesis, GLP-1 peptide mimetics or DPP-IV inhibitors could exert oncogenic effects [116]. Based on the lack of detection of GLP-1 receptors in 21 human pancreatic adenocarcinomas, Korner et al. [117] suggested that GLP-1 expression could be restricted to certain cell lines and, therefore, could be irrelevant in humans. Moreover, exantine, a GLP-1 analogue, neither modulated the growth of pancreatic cancer cells which expressed the receptor, nor rescued them from drug-induced death. Furthermore, the sustained exantine-mediated activation of the receptor did not stimulate tumor growth or progression in rats [118]. These observations suggested that the hypothetical appearance of prostate cancer by administering DPP-IV inhibitors or GLP-1 mimetics could be caused by collateral pancreatitis [116], a condition favored by underlying diseases such as type II diabetes and obesity [119]. In fact, pancreatitis incidence in patients treated with GLP-1 receptor agonists does not differ with that found in populations suffering from type II diabetes [120, 121]. Similarly, studies in rats and monkeys indicated that the induction of pancreatitis through the stimulation of the GLP-1 receptor seems to be quite improbable [122].
Preclinical studies have shown an increased incidence of thyroid C-cell tumors in rodents treated with GLP-1 analogues. Nevertheless, the GLP-1 receptor expression in this cell type is highly dependent on the species, implying that observations in rodents are not necessarily relevant in human [117] due to a differential expression 22-times higher in rodents [123, 124]. Besides, it was shown that rats are more susceptible to develop thyroid C-cell neoplasias, quite rare in humans [125].
The rise in GLP-1 concentrations seems to originate more probably from premalignant lesions stimulation rather than the induction of new lesions [117], both in pancreas and the thyroid, based on the short duration of the studies and the evidences gathered so far. In contrast, it is believed that GLP-1 receptor activation could inhibit tumor growth in two very common cancer types: colon and breast cancers [117].
In the case of colon cancer, the CT26 murine colon cancer cell line expresses a functional GLP-1 receptor. Its exposure to exentanide in vitro leads to morphological changes, inhibits proliferation and colony formation in solid agar and induces apoptosis. These effects were confirmed in vivo in CT26 cells implanted in mice, even when this did not affect tumor weight [126].
Ligumsky et al., [127] demonstrated in vitro that exentanide significantly reduced the number of colonies formed by the cell lines MCF-1 and MDA-MB-231, positive and negative to estrogen receptors, respectively. The non-cancer HB-2 cell line remained unaffected. A significant exentanide dose-dependent tumor reduction was seen in mice implanted with MDA-MB-231 cells, when the drug was administered by the intraperitoneal route [128].
CONCLUSIONS
Currently, DPP-IV gets the attention of the international scientific community, due to its peculiarly complex tridimensional structure. This feature determines DPP-IV molecular and functional properties, and its role in both the physiological and pathological processes mediated by its enzyme activity or its interaction with other proteins. It is implicated in mammalian homeostasis maintenance and, significantly, in the molecular mechanisms of multiple diseases, particularly cancer and immunological disorders, where it is found to have altered expression or misbalanced activity. A better comprehension on the role of DPP-IV in those disease-related processes would make it a very attractive target to design and develop more effective therapeutic strategies.
ACKNOWLEDGEMENTS
The authors thanks the Project 3276/3 of the International Foundation for Science (IFS), together with the Chemical Weapons Prohibition Organization, for the financial support.
REFERENCES
1. Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5(9):785-99.
2. Leung D, Abbenante G, Fairlie DP. Protease inhibitors: current status and future prospects. J Med Chem. 2000;43(3):305-41.
3. Abbenante G, Fairlie DP. Protease inhibitors in the clinic. Med Chem. 2005;1(1):71-104.
4. Itou M, Kawaguchi T, Taniguchi E, Sata M. Dipeptidyl peptidase-4: a key player in chronic liver disease. World J Gastroenterol. 2013;19(15):2298-306.
5. Gorrell MD, Wang XM, Park J, Ajami K, Yu DM, Knott H, et al. Structure and function in dipeptidyl peptidase IV and related proteins. Adv Exp Med Biol. 2006;575:45-54.
6. Yu DM, Yao TW, Chowdhury S, Nadvi NA, Osborne B, Church WB, et al. The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J. 2010;277(5):1126-44.
7. Leiting B, Pryor KD, Wu JK, Marsilio F, Patel RA, Craik CS, et al. Catalytic properties and inhibition of proline-specific dipeptidyl peptidases II, IV and VII. Biochem J. 2003;371(Pt 2):525-32.
8. Hopsu-Havu VK, Glenner GG. A new dipeptide naphthylamidase hydrolyzing glycyl-prolyl-beta-naphthylamide. Histochemie. 1966;7(3):197-201.
9. Palmieri FE, Ward PE. Dipeptidyl(amino)peptidase IV and post proline cleaving enzyme in cultured endothelial and smooth muscle cells. Adv Exp Med Biol. 1989;247A:305-11.
10. Hu CX, Huang H, Zhang L, Huang Y, Shen ZF, Cheng KD, et al. A new screening method based on yeast-expressed human dipeptidyl peptidase IV and discovery of novel inhibitors. Biotechnol Lett. 2009;31(7):979-84.
11. Durinx C, Lambeir AM, Bosmans E, Falmagne JB, Berghmans R, Haemers A, et al. Molecular characterization of dipeptidyl peptidase activity in serum: soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur J Biochem. 2000;267(17):5608-13.
12. Stano J, Kovacs P, Kakoniova D, Kirilova ND, Komov VP. Activity of dipeptidyl peptidase IV in gingseng callus culture. Biologia. 1994;49:353-7.
13. Koreeda Y, Hayakawa M, Ikemi T, Abiko Y. Isolation and characterisation of dipeptidyl peptidase IV from Prevotella loescheii ATCC 15930. Arch Oral Biol. 2001;46(8):759-66.
14. Davy A, Thomsen KK, Juliano MA, Alves LC, Svendsen I, Simpson DJ. Purification and characterization of barley dipeptidyl peptidase IV. Plant Physiol. 2000;122(2):425-32.
15. Bauvois B, Djavaheri-Mergny M, Rouillard D, Dumont J, Wietzerbin J. Regulation of CD26/DPPIV gene expression by interferons and retinoic acid in tumor B cells. Oncogene. 2000;19(2):265-72.
16. de Meester I, Vanhoof G, Lambeir AM, Scharpe S. Use of immobilized adenosine deaminase (EC 3.5.4.4) for the rapid purification of native human CD26/dipeptidyl peptidase IV (EC 3.4.14.5). J Immunol Methods. 1996;189(1):99-105.
17. Wilson MJ, Ruhland AR, Pryor JL, Ercole C, Sinha AA, Hensleigh H, et al. Prostate specific origin of dipeptidylpeptidase IV (CD-26) in human seminal plasma. J Urol. 1998;160(5):1905-9.
18. Engel M, Hoffmann T, Wagner L, Wermann M, Heiser U, Kiefersauer R, et al. The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc Natl Acad Sci USA. 2003;100(9):5063-8.
19. Abbott CA, Baker E, Sutherland GR, McCaughan GW. Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics. 1994;40(5):331-8.
20. Bohm SK, Gum JR, Jr., Erickson RH, Hicks JW, Kim YS. Human dipeptidyl peptidase IV gene promoter: tissue-specific regulation from a TATA-less GC-rich sequence characteristic of a housekeeping gene promoter. Biochem J. 1995;311 ( Pt 3):835-43.
21. Hong WJ, Petell JK, Swank D, Sanford J, Hixson DC, Doyle D. Expression of dipeptidyl peptidase IV in rat tissues is mainly regulated at the mRNA levels. Exp Cell Res. 1989;182(1):256-66.
22. Kahne T, Kroning H, Thiel U, Ulmer AJ, Flad HD, Ansorge S. Alterations in structure and cellular localization of molecular forms of DP IV/CD26 during T cell activation. Cell Immunol. 1996;170(1):63-70.
23. Gorrell MD. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin Sci (Lond). 2005;108(4):277-92.
24. Yu DM, Ajami K, Gall MG, Park J, Lee CS, Evans KA, et al. The in vivo expression of dipeptidyl peptidases 8 and 9. J Histochem Cytochem. 2009;57(11):1025-40.
25. Lee KN, Jackson KW, Christiansen VJ, Chung KH, McKee PA. A novel plasma proteinase potentiates alpha2-antiplasmin inhibition of fibrin digestion. Blood. 2004;103(10):3783-8.
26. Ajami K, Abbott CA, McCaughan GW, Gorrell MD. Dipeptidyl peptidase 9 has two forms, a broad tissue distribution, cytoplasmic localization and DPIV-like peptidase activity. Biochim Biophys Acta. 2004;1679(1):18-28.
27. Delacour D, Gouyer V, Leteurtre E, Ait-Slimane T, Drobecq H, Lenoir C, et al. 1-benzyl-2-acetamido-2-deoxy-alpha-D-galactopyranoside blocks the apical biosynthetic pathway in polarized HT-29 cells. J Biol Chem. 2003;278(39):37799-809.
28. Rasmussen HB, Branner S, Wiberg FC, Wagtmann N. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat Struct Biol. 2003;10(1):19-25.
29. Duke-Cohan JS, Morimoto C, Rocker JA, Schlossman SF. Serum high molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen DPPT-L released from activated T cells. J Immunol. 1996;156(5):1714-21.
30. Pascual I, Gomez H, Pons T, Chappe M, Vargas MA, Valdes G, et al. Effect of divalent cations on the porcine kidney cortex membrane-bound form of dipeptidyl peptidase IV. Int J Biochem Cell Biol. 2011;43(3):363-71.
31. Gomez H, Chappe M, Valiente PA, Pons T, Chavez Mde L, Charli JL, et al. Effect of zinc and calcium ions on the rat kidney membrane-bound form of dipeptidyl peptidase IV. J Biosci. 2013;38(3):461-9.
32. Thoma R, Loffler B, Stihle M, Huber W, Ruf A, Hennig M. Structural basis of proline-specific exopeptidase activity as observed in human dipeptidyl peptidase-IV. Structure. 2003;11(8):947-59.
33. Aertgeerts K, Ye S, Tennant MG, Kraus ML, Rogers J, Sang BC, et al. Crystal structure of human dipeptidyl peptidase IV in complex with a decapeptide reveals details on substrate specificity and tetrahedral intermediate formation. Protein Sci. 2004;13(2):412-21.
34. Abbott CA, McCaughan GW, Gorrell MD. Two highly conserved glutamic acid residues in the predicted beta propeller domain of dipeptidyl peptidase IV are required for its enzyme activity. FEBS Lett. 1999;458(3):278-84.
35. Ajami K, Abbott CA, Obradovic M, Gysbers V, Kahne T, McCaughan GW, et al. Structural requirements for catalysis, expression, and dimerization in the CD26/DPIV gene family. Biochemistry. 2003;42(3):694-701.
36. Chien CH, Huang LH, Chou CY, Chen YS, Han YS, Chang GG, et al. One site mutation disrupts dimer formation in human DPP-IV proteins. J Biol Chem. 2004;279(50):52338-45.
37. Varghese JN, Laver WG, Colman PM. Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 A resolution. Nature. 1983;303(5912):35-40.
38. Vellieux FM, Huitema F, Groendijk H, Kalk KH, Jzn JF, Jongejan JA, et al. Structure of quinoprotein methylamine dehydrogenase at 2.25 A resolution. EMBO J. 1989;8(8):2171-8.
39. Ito N, Phillips SE, Stevens C, Ogel ZB, McPherson MJ, Keen JN, et al. Novel thioether bond revealed by a 1.7 A crystal structure of galactose oxidase. Nature. 1991;350(6313):87-90.
40. Xia ZX, Dai WW, Xiong JP, Hao ZP, Davidson VL, White S, et al. The three-dimensional structures of methanol dehydrogenase from two methylotrophic bacteria at 2.6-A resolution. J Biol Chem. 1992;267(31):22289-97.
41. Murzin AG. Structural principles for the propeller assembly of beta-sheets: the preference for seven-fold symmetry. Proteins. 1992;14(2):191-201.
42. Fülöp V, Jones DT. Beta propellers: structural rigidity and functional diversity. Curr Opin Struct Biol. 1999;9(6):715-21.
43. Paoli M. Protein folds propelled by diversity. Prog Biophys Mol Biol. 2001;76(1-2):103-30.
44. Jawad Z, Paoli M. Novel sequences propel familiar folds. Structure. 2002;10(4):447-54.
45. Adams J, Kelso R, Cooley L. The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol. 2000;10(1):17-24.
46. Russell RB, Sasieni PD, Sternberg MJ. Supersites within superfolds. Binding site similarity in the absence of homology. J Mol Biol. 1998;282(4):903-18.
47. Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307(4):1113-43.
48. Pons T, Gomez R, Chinea G, Valencia A. Beta-propellers: associated functions and their role in human diseases. Curr Med Chem. 2003;10(6):505-24.
49. Love CA, Harlos K, Mavaddat N, Davis SJ, Stuart DI, Jones EY, et al. The ligand-binding face of the semaphorins revealed by the high-resolution crystal structure of SEMA4D. Nat Struct Biol. 2003;10(10):843-8.
50. Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294(5541):339-45.
51. Hiramatsu H, Yamamoto A, Kyono K, Higashiyama Y, Fukushima C, Shima H, et al. The crystal structure of human dipeptidyl peptidase IV (DPPIV) complex with diprotin A. Biol Chem. 2004;385(6):561-4.
52. Oefner C, D'Arcy A, Mac Sweeney A, Pierau S, Gardiner R, Dale GE. High-resolution structure of human apo dipeptidyl peptidase IV/CD26 and its complex with 1-[([2-[(5-iodopyridin-2-yl)amino]-ethyl]amino)-acetyl]-2-cyano-(S)-pyrrolidine. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 7):1206-12.
53. Weihofen WA, Liu J, Reutter W, Saenger W, Fan H. Crystal structure of CD26/dipeptidyl-peptidase IV in complex with adenosine deaminase reveals a highly amphiphilic interface. J Biol Chem. 2004;279(41):43330-5.
54. Abbott CA, Gorrell MD. The family of CD26/DPP-IV and related ectopeptidases. In: Langner J, Ansorge S, editors. Ectopeptidases. CD13/aminopeptidase N and CD26/dipeptidylpeptidase IV in medicine and biology. New York: Kluwer Academic / Plenum Publishers; 2002. p. 171-95.
55. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57-70.
56. Carl-McGrath S, Lendeckel U, Ebert M, Rocken C. Ectopeptidases in tumour biology: a review. Histol Histopathol. 2006;21(12):1339-53.
57. Iwata S, Morimoto C. CD26/dipeptidyl peptidase IV in context. The different roles of a multifunctional ectoenzyme in malignant transformation. J Exp Med. 1999;190(3):301-6.
58. Wesley UV, Albino AP, Tiwari S, Houghton AN. A role for dipeptidyl peptidase IV in suppressing the malignant phenotype of melanocytic cells. J Exp Med. 1999;190(3):311-22.
59. Sedo A, Krepela E, Kasafirek E. Dipeptidyl peptidase IV, prolyl endopeptidase and cathepsin B activities in primary human lung tumors and lung parenchyma. J Cancer Res Clin Oncol. 1991;117(3):249-53.
60. Asada Y, Aratake Y, Kotani T, Marutsuka K, Araki Y, Ohtaki S, et al. Expression of dipeptidyl aminopeptidase IV activity in human lung carcinoma. Histopathology. 1993;23(3):265-70.
61. Wesley UV, Tiwari S, Houghton AN. Role for dipeptidyl peptidase IV in tumor suppression of human non small cell lung carcinoma cells. Int J Cancer. 2004;109(6):855-66.
62. Dimitrova M, Ivanov I, Todorova R, Stefanova N, Moskova-Doumanova V, Topouzova-Hristova T, et al. Comparison of the activity levels and localization of dipeptidyl peptidase IV in normal and tumor human lung cells. Tissue Cell. 2012;44(2):74-9.
63. Bogenrieder T, Finstad CL, Freeman RH, Papandreou CN, Scher HI, Albino AP, et al. Expression and localization of aminopeptidase A, aminopeptidase N, and dipeptidyl peptidase IV in benign and malignant human prostate tissue. Prostate. 1997;33(4):225-32.
64. Frohlich E, Maier E, Wahl R. Interspecies differences in membrane-associated protease activities of thyrocytes and their relevance for thyroid cancer studies. J Exp Clin Cancer Res. 2012;31:45.
65. Kotani T, Asada Y, Aratake Y, Umeki K, Yamamoto I, Tokudome R, et al. Diagnostic usefulness of dipeptidyl aminopeptidase IV monoclonal antibody in paraffin-embedded thyroid follicular tumours. J Pathol. 1992;168(1):41-5.
66. Tanaka T, Umeki K, Yamamoto I, Sakamoto F, Noguchi S, Ohtaki S. CD26 (dipeptidyl peptidase IV/DPP IV) as a novel molecular marker for differentiated thyroid carcinoma. Int J Cancer. 1995;64(5):326-31.
67. Tang AC, Raphael SJ, Lampe HB, Matthews TW, Becks GP. Expression of dipeptidyl aminopeptidase IV activity in thyroid tumours: a possible marker of thyroid malignancy. J Otolaryngol. 1996;25(1):14-9.
68. Maruta J, Hashimoto H, Yamashita H, Yamashita H, Noguchi S. Diagnostic applicability of dipeptidyl aminopeptidase IV activity in cytological samples for differentiating follicular thyroid carcinoma from follicular adenoma. Arch Surg. 2004;139(1):83-8.
69. Khin EE, Kikkawa F, Ino K, Kajiyama H, Suzuki T, Shibata K, et al. Dipeptidyl peptidase IV expression in endometrial endometrioid adenocarcinoma and its inverse correlation with tumor grade. Am J Obstet Gynecol. 2003;188(3):670-6.
70. Kikkawa F, Kajiyama H, Ino K, Shibata K, Mizutani S. Increased adhesion potency of ovarian carcinoma cells to mesothelial cells by overexpression of dipeptidyl peptidase IV. Int J Cancer. 2003;105(6):779-83.
71. Pro B, Dang NH. CD26/dipeptidyl peptidase IV and its role in cancer. Histol Histopathol. 2004;19(4):1345-51.
72. Goscinski MA, Suo ZH, Nesland JM, Florenes VA, Giercksky KE. Dipeptidyl peptidase IV expression in cancer and stromal cells of human esophageal squamous cell carcinomas, adenocarcinomas and squamous cell carcinoma cell lines. APMIS. 2008;116(9):823-31.
73. Bauvois B, De Meester I, Dumont J, Rouillard D, Zhao HX, Bosmans E. Constitutive expression of CD26/dipeptidylpeptidase IV on peripheral blood B lymphocytes of patients with B chronic lymphocytic leukaemia. Br J Cancer. 1999;79(7-8):1042-8.
74. Cro L, Morabito F, Zucal N, Fabris S, Lionetti M, Cutrona G, et al. CD26 expression in mature B-cell neoplasia: its possible role as a new prognostic marker in B-CLL. Hematol Oncol. 2009;27(3):140-7.
75. Havre PA, Dang LH, Ohnuma K, Iwata S, Morimoto C, Dang NH. CD26 expression on T-anaplastic large cell lymphoma (ALCL) line Karpas 299 is associated with increased expression of versican and MT1-MMP and enhanced adhesion. BMC cancer. 2013;13:517.
76. Stremenova J, Krepela E, Mares V, Trim J, Dbaly V, Marek J, et al. Expression and enzymatic activity of dipeptidyl peptidase-IV in human astrocytic tumours are associated with tumour grade. Int J Oncol. 2007;31(4):785-92.
77. Stremenova J, Mares V, Lisa V, Hilser M, Krepela E, Vanickova Z, et al. Expression of dipeptidyl peptidase-IV activity and/or structure homologs in human meningiomas. Int J Oncol. 2010;36(2):351-8.
78. Arscott WT, LaBauve AE, May V, Wesley UV. Suppression of neuroblastoma growth by dipeptidyl peptidase IV: relevance of chemokine regulation and caspase activation. Oncogene. 2009;28(4):479-91.
79. Dinjens WN, Ten Kate J, Kirch JA, Tanke HJ, Van der Linden EP, Van den Ingh HF, et al. Adenosine deaminase complexing protein (ADCP) expression and metastatic potential in prostatic adenocarcinomas. J Pathol. 1990;160(3):195-201.
80. Wesley UV, McGroarty M, Homoyouni A. Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res. 2005;65(4):1325-34.
81. Urade M, Komatsu M, Yamaoka M, Fukasawa K, Harada M, Mima T, et al. Serum dipeptidyl peptidase activities as a possible marker of oral cancer. Cancer. 1989;64(6):1274-80.
82. de la Haba-Rodriguez J, Macho A, Calzado MA, Blazquez MV, Gomez MA, Munoz EE, et al. Soluble dipeptidyl peptidase IV (CD-26) in serum of patients with colorectal carcinoma. Neoplasma. 2002;49(5):307-11.
83. Cordero OJ, Imbernon M, Chiara LD, Martinez-Zorzano VS, Ayude D, de la Cadena MP, et al. Potential of soluble CD26 as a serum marker for colorectal cancer detection. World J Clin Oncol. 2011;2(6):245-61.
84. Gonzalez-Gronow M, Misra UK, Gawdi G, Pizzo SV. Association of plasminogen with dipeptidyl peptidase IV and Na+/H+ exchanger isoform NHE3 regulates invasion of human 1-LN prostate tumor cells. J Biol Chem. 2005;280(29):27173-8.
85. Gorrell MD, Gysbers V, McCaughan GW. CD26: a multifunctional integral membrane and secreted protein of activated lymphocytes. Scand J Immunol. 2001;54(3):249-64.
86. Cheng HC, Abdel-Ghany M, Pauli BU. A novel consensus motif in fibronectin mediates dipeptidyl peptidase IV adhesion and metastasis. J Biol Chem. 2003;278(27):24600-7.
87. Cheng HC, Abdel-Ghany M, Elble RC, Pauli BU. Lung endothelial dipeptidyl peptidase IV promotes adhesion and metastasis of rat breast cancer cells via tumor cell surface-associated fibronectin. J Biol Chem. 1998;273(37):24207-15.
88. Korach S, Poupon MF, Du Villard JA, Becker M. Differential adhesiveness of rhabdomyosarcoma-derived cloned metastatic cell lines to vascular endothelial monolayers. Cancer Res. 1986;46(7):3624-9.
89. Kato Y, Saijo N. Developed new agents for lung cancer. Nihon Geka Gakkai zasshi. 2002;103(2):218-23.
90. Mentlein R, Dahms P, Grandt D, Kruger R. Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regul Pept. 1993;49(2):133-44.
91. Ghersi G, Chen W, Lee EW, Zukowska Z. Critical role of dipeptidyl peptidase IV in neuropeptide Y-mediated endothelial cell migration in response to wounding. Peptides. 2001;22(3):453-8.
92. Mathew S, Morrison ME, Murty VV, Houghton AN, Chaganti RS. Assignment of the DPP4 gene encoding adenosine deaminase binding protein (CD26/dipeptidylpeptidase IV) to 2q23. Genomics. 1994;22(1):211-2.
93. Otsuka T, Kohno T, Mori M, Noguchi M, Hirohashi S, Yokota J. Deletion mapping of chromosome 2 in human lung carcinoma. Genes Chromosomes Cancer. 1996;16(2):113-9.
94. Shiseki M, Kohno T, Nishikawa R, Sameshima Y, Mizoguchi H, Yokota J. Frequent allelic losses on chromosomes 2q, 18q, and 22q in advanced non-small cell lung carcinoma. Cancer Res. 1994;54(21):5643-8.
95. Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001;15(8):968-80.
96. Yan P, Muhlethaler A, Bourloud KB, Beck MN, Gross N. Hypermethylation-mediated regulation of CD44 gene expression in human neuroblastoma. Genes Chromosomes Cancer. 2003;36(2):129-38.
97. Kajiyama H, Kikkawa F, Maeda O, Suzuki T, Ino K, Mizutani S. Increased expression of dipeptidyl peptidase IV in human mesothelial cells by malignant ascites from ovarian carcinoma patients. Oncology. 2002;63(2):158-65.
98. Ware JL. Growth factor network disruption in prostate cancer progression. Cancer Metastasis Rev. 1998;17(4):443-7.
99. Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res. 1999;5(5):1063-71.
100. Dow JK, deVere White RW. Fibroblast growth factor 2: its structure and property, paracrine function, tumor angiogenesis, and prostate-related mitogenic and oncogenic functions. Urology. 2000;55(6):800-6.
101. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1(1):34-45.
102. Isaacs JT, Isaacs WB. Androgen receptor outwits prostate cancer drugs. Nat Med. 2004;10(1):26-7.
103. Boonacker E, Van Noorden CJ. The multifunctional or moonlighting protein CD26/DPPIV. Eur J Cell Biol. 2003;82(2):53-73.
104. Proost P, Struyf S, Schols D, Opdenakker G, Sozzani S, Allavena P, et al. Truncation of macrophage-derived chemokine by CD26/ dipeptidyl-peptidase IV beyond its predicted cleavage site affects chemotactic activity and CC chemokine receptor 4 interaction. J Biol Chem. 1999;274(7):3988-93.
105. Nakamoto T, Chang CS, Li AK, Chodak GW. Basic fibroblast growth factor in human prostate cancer cells. Cancer Res. 1992;52(3):571-7.
106. Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr., Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999;59(2):279-84.
107. Pintucci G, Moscatelli D, Saponara F, Biernacki PR, Baumann FG, Bizekis C, et al. Lack of ERK activation and cell migration in FGF-2-deficient endothelial cells. FASEB J. 2002;16(6):598-600.
108. Giuliani R, Bastaki M, Coltrini D, Presta M. Role of endothelial cell extracellular signal-regulated kinase1/2 in urokinase-type plasminogen activator upregulation and in vitro angiogenesis by fibroblast growth factor-2. J Cell Sci. 1999;112 (Pt 15):2597-606.
109. Rabbani SA, Mazar AP. The role of the plasminogen activation system in angiogenesis and metastasis. Surg Oncol Clin N Am. 2001;10(2):393-415.
110. Bugler B, Amalric F, Prats H. Alternative initiation of translation determines cytoplasmic or nuclear localization of basic fibroblast growth factor. Mol Cell Biol. 1991;11(1):573-7.
111. Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18(1):26-45.
112. Delrieu I. The high molecular weight isoforms of basic fibroblast growth factor (FGF-2): an insight into an intracrine mechanism. FEBS Lett. 2000;468(1):6-10.
113. Arwert EN, Mentink RA, Driskell RR, Hoste E, Goldie SJ, Quist S, et al. Upregulation of CD26 expression in epithelial cells and stromal cells during wound-induced skin tumour formation. Oncogene. 2012;31(8):992-1000.
114. Ehtesham M, Winston JA, Kabos P, Thompson RC. CXCR4 expression mediates glioma cell invasiveness. Oncogene. 2006;25(19):2801-6.
115. Christopherson KW, 2nd, Hangoc G, Broxmeyer HE. Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1 alpha-mediated chemotaxis of human cord blood CD34+ progenitor cells. J Immunol. 2002;169(12):7000-8.
116. Vangoitsenhoven R, Mathieu C, Van der Schueren B. GLP1 and cancer: friend or foe? Endocr Relat Cancer. 2012;19(5):F77-88.
117. Korner M, Stockli M, Waser B, Reubi JC. GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med. 2007;48(5):736-43.
118. Koehler JA, Drucker DJ. Activation of glucagon-like peptide-1 receptor signaling does not modify the growth or apoptosis of human pancreatic cancer cells. Diabetes. 2006;55(5):1369-79.
119. Girman CJ, Kou TD, Cai B, Alexander CM, O'Neill EA, Williams-Herman DE, et al. Patients with type 2 diabetes mellitus have higher risk for acute pancreatitis compared with those without diabetes. Diabetes Obes Metab. 2010;12(9):766-71.
120. Garg R, Chen W, Pendergrass M. Acute pancreatitis in type 2 diabetes treated with exenatide or sitagliptin: a retrospective observational pharmacy claims analysis. Diabetes Care. 2010;33(11):2349-54.
121. Dore DD, Bloomgren GL, Wenten M, Hoffman C, Clifford CR, Quinn SG, et al. A cohort study of acute pancreatitis in relation to exenatide use. Diabetes Obes Metab. 2011;13(6):559-66.
122. Nyborg NC, Molck AM, Madsen LW, Knudsen LB. The human GLP-1 analog liraglutide and the pancreas: evidence for the absence of structural pancreatic changes in three species. Diabetes. 2012;61(5):1243-9.
123. Bjerre Knudsen L, Madsen LW, Andersen S, Almholt K, de Boer AS, Drucker DJ, et al. Glucagon-like Peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology. 2010;151(4):1473-86.
124. Waser B, Beetschen K, Pellegata NS, Reubi JC. Incretin receptors in non-neoplastic and neoplastic thyroid C cells in rodents and humans: relevance for incretin-based diabetes therapy. Neuroendocrinology. 2011;94(4):291-301.
125. Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006;107(9):2134-42.
126. Koehler JA, Kain T, Drucker DJ. Glucagon-like peptide-1 receptor activation inhibits growth and augments apoptosis in murine CT26 colon cancer cells. Endocrinology. 2011;152(9):3362-72.
127. Ligumsky H, Wolf I, Israeli S, Haimsohn M, Ferber S, Karasik A, et al. The peptide-hormone glucagon-like peptide-1 activates cAMP and inhibits growth of breast cancer cells. Breast Cancer Res Treat. 2012;132(2):449-61.
128. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326-31.
Received in September, 2013.
Accepted in April, 2014.
Isel Pascual. Centro de Estudios de Proteínas (CEP), Facultad de Biología, Universidad de La Habana, UH. Calle 25 No. 455, Vedado, CP 10400, La Habana, Cuba. E-mail: isel@fbio.uh.cu; iselpascual@yahoo.es.

{kind=link}
{kind=link}
{kind=link}