










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.31 no.4 La Habana Oct.-Dec. 2014
RESEARCH
High glucose burden inhibits EGFR/PI3K/AKT1/mTOR signaling pathway in cutaneous fibroblasts
Altas concentraciones de glucosa inhiben la cascada de señalización EGFR/PI3K/AKT1/mTOR en fibroblastos cutáneos
Yssel Mendoza-Marí1, Ariana Garcia-Ojalvo1, Maday Fernández-Mayola1, Luis Herrera-Martínez2, Jorge Berlanga-Acosta1
1 Tissue Repair and Cyto-protection Laboratory, Direction of Biomedical Research. Center for Genetic Engineering and Biotechnology, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.
2 General Direction. Center for Genetic Engineering and Biotechnology, CIGB. La Habana, Cuba.
ABSTRACT
Diabetic healing failure is the clinical expression of countless molecular and cellular disorders having hyperglycemia as the proximal trigger. The fibroblast is a critical building-block cell type for the healing process. Under high glucose concentrations fibroblasts physiology is perturbed. The epidermal growth factor receptor (EGFR) signaling system is crucial for different healing events. We examined the effect of high glucose burden on healthy-donor’ cutaneous fibroblasts proliferation, so as the EGFR autophosphorylation on a critical tyrosine residue, along with the activation of downstream signaling pathways proximal to cyclin D1 expression. Fibroblasts were cultured in 15 % FBS at either 5 (normal glucose) or 35 (high glucose) mM under standard culture conditions. Concurrent osmotic control was included. After 6 days of incubation under high glucose, doubling time was calculated. Cells suspensions were plated, fixed and immunolabelled with antibodies directed to phosphorylated forms of EGFR (Y1197), AKT1 (S473) and mTOR (S2448), and native forms of PI3K p85 alpha subunit and cyclin D1. The ratio of cells positive to the diaminobenzidine/peroxidase reaction was calculated and its intensity estimated according to published methodologies. High glucose concentration significantly increased doubling time 5-fold, as compared to cells grown in physiological conditions. Hyperglycemia reduced the constitutive EGFR autophosphorylation. Accordingly, PI3K was also significantly attenuated. Downstream switches AKT1 and mTOR were also affected and very significantly on the signal intensity. Cyclin D1 expression was completely abrogated due to high glucose burden. Collectively, these data suggest that high glucose exposure hinders fibroblasts proliferation by disrupting the EGFR/PI3K/AKT1/mTOR/Cyclin D1 axis.
Keywords: fibroblasts, hyperglycemia, diabetic ulcer, signaling pathways, EGFR.
RESUMEN
La fisiología de los fibroblastos, esenciales para la cicatrización, se perturba ante altas concentraciones de glucosa. Se estudió el efecto de altas cargas de glucosa sobre la proliferación de fibroblastos cutáneos de donantes sanos a través de la cascada de señalización del factor de crecimiento epidérmico (EGFR): la autofosforilación del EGFR y la activación de intermediarios de la transducción hasta la ciclina D1. Los fibroblastos se cultivaron durante seis días en condiciones estándar con SFB al 15 %, y glucosa a 5 mM (glucosa normal) o 35 mM (glucosa alta). Se incluyó un control osmótico concurrente. Tras seis días se estimó el tiempo de duplicación. Las suspensiones celulares se sembraron, fijaron e inmunomarcaron con anticuerpos específicos contra las formas fosforiladas de EGFR (Y1197), AKT1 (S473) y mTOR (S2448), y las formas nativas de la subunidad p85 alfa de la PI3K y la ciclina D1. Se calculó la proporción de células positivas a la reacción de diaminobenzidina/peroxidasa, y su intensidad según procedimientos estándar. Las altas concentraciones de glucosa multiplicaron en 5 veces el tiempo de duplicación comparado con el de células cultivadas en condiciones fisiológicas. También redujo la autofosforilación constitutiva del EGFR, atenuó significativamente la subsiguiente fosforilación de PI3K y muy significativamente la de sus intermediarios de transducción AKT1 y mTOR. La expresión de la ciclina D1 se abolió ante las altas cargas de glucosa. Los resultados sugieren que la exposición a altas concentraciones de glucosa afecta la proliferación de los fibroblastos al perturbar el eje de transducción EGFR/PI3K/AKT1/mTOR/Ciclina D1.
Palabras clave: fibroblastos, hiperglucemia, úlcera diabética, cascadas de señalización, EGFR.
INTRODUCTION
Diabetes mellitus type 2 (DM2) represents a worldwide pandemic disease [1]. In this context, wound healing failure is a significant complication of DM associated with increased disability, morbidity and mortality due to lower limb ulcerations and the likelihood of amputation [2-6]. Hyperglycemia and its derived biochemical disturbances undermine the whole tissues’ functioning and structure, orchestrating irreversible systemic complications from which the skin and soft peripheral tissue cells do not escape [7]. Thus, diabetic wound healing failure is the clinical gross expression of an outstanding array of subtle molecular and cellular disorders [8].
Fibroblasts are an essential cell type for the wound healing process, by secreting, contracting and remodeling the extracellular matrix (ECM). One of their main functions is to secrete growth factors as vital messengers for mesenchymal and epithelial communication, especially for establishing the emerging basement membrane and the subsequent epithelial migration [9]. Consequently, any impediment to fibroblast functioning is deterrent for normal wound healing and may lead to wound chronification.
Under the high glucose burden imposed by DM, fibroblasts appear perturbed and for many years, in vitro models recreating “clinical hyperglycemia” have proved to disrupt normal fibroblasts physiology and derange the secretion of extracellular matrix ingredients. These experiments have suggested that acute and chronic exposure to high glucose concentration is the proximal toxic trigger for cutaneous fibroblasts’ demise [10-15]. Nevertheless, the molecular mechanisms underlying the glucotoxicity-associated fibroblasts’ proliferation reluctance and premature senescence have not been enlightened.
Additionally, interfering epidermal growth factor receptor (EGFR) cascade acts as a restrictive factor for normal tissue repair by reducing cell proliferation among other factors [16]. Portero and co-workers successfully demonstrated that one of the advanced glycation end-products (AGEs) precursors, and vastly represented in diabetic patients circulation, abrogates EGFR tyrosine autophosphorylation and the subsequent activation of diverse downstream signaling kinases [17]. These findings encouraged us to examine the potential impact of a high glucose burden on the EGFR and downstream-related kinases phosphorylation, using primarily cultured human cutaneous fibroblasts.
Here we provide the first evidences that the exposure to a high glucose burden reduces the autophosphorylation ability of a catalytic tyrosine residue of the EGFR in dermal fibroblasts. Accordingly, subsequent underphosphorylation on downstream signaling substrates is detected along with an eventual suppression of cyclin D1 expression. These evidences provide a theoretical frame to explain, at least in part, the cytotoxic effects of high glucose on granulation tissue-productive cells.
MATERIALS AND METHODS
Ethics statement
Pieces of human foreskin were obtained from circumcision of a human infant, who underwent mandatory surgical procedure at the Juan Manuel Márquez Pediatrics Hospital, Havana, Cuba. Written consent was obtained from the donor’s parents approving the use of child’s removed tissue for experimentation. Protocols were approved by the Ethics Committee of the Juan Manuel Márquez Pediatric Hospital.
Cutaneous fibroblasts culture
Tissue fragments were placed immediately in serum-free Dulbecco’s modified Eagles medium (DMEM)/ Ham’s F12 culture medium (1:1), and conserved at 4 °C for transport to the laboratory. The samples were subdivided into pieces of 1–2 mm3, orderly placed in 60 mm culture plates and incubated for 2 h at 37 °C in 5 % CO2 atmosphere. Subsequently, low glucose (1 g/L, equivalent to 5.5 mM) DMEM supplemented with 30 % fetal bovine serum (FBS), 1× antibiotic -antimycotic solution, 2 mM glutamine, was added slowly to avoid tissue fragments detachment. Once fibroblasts sprouts started to emerge from each explant, cells were fed every 4-5 days and maintained in a humidified, 5 % CO2 atmosphere at 37 °C. The experiments described here were conducted with cells from healthy infants of non-diabetic parental background and under passages 3-5.
Hyperglycemia stress
Three independent and extemporaneous experiments were conducted under the same stressor conditions with cells on passages 3-5. When reaching approximately 90 % confluence, cultures were trypsinized and an homogeneous number of cells was plated in p60 dishes with DMEM devoid of FBS for 48 h to synchronize cells’ cycle. Afterwards, medium was replaced and replenished with 15 % FBS under the following culture conditions: D-glucose at either 5 mM (normal glucose) or 35 mM (high glucose). Concurrent osmotic control consisted in fibroblasts exposed to 35 mM glucose as largely used [18]. After 6 days of “chronic” exposure to this high glucose burden, cells were quantified according to Trypan blue dying exclusion method and doubling time was calculated as described [19]. Cell suspensions adjusted to 3000 cells/25 μL were plated onto sterile silanized slides (Dako, USA) in the same culture medium and incubated overnight for another 24 h in moist chamber to ensure attachment. Afterwards, the slides were washed in cold phosphate-buffered saline solution (pH 7.2; PBS), immersed in ice cold acetone/methanol 1:1 (v/v) for 10 min and air-dried at room temperature.
Immunocytochemistry
As the main goal of this experiment was to learn on the effect of high glucose concentration exposure on the EGFR and key signaling substrates, five antibodies were selected to study their posttranslational modification (Table). They were purchased from Abcam (USA) and used at a 1:100 dilution, in a commercial antibody diluent solution (Abcam 64211).
The immunocytochemistry reaction was developed following the manufacturer’s instructions of a commercial anti-mouse & anti-rabbit HRP/DAB detection kit (Abcam 64264). Briefly, slides were washed in PBS for 15 min and cells permeabilized by immersion in PBS-Tween (0.1 % Tween 20) for 2 min at room temperature, and washed three times for 5 min with PBS. Endogenous peroxidase was quenched with the Dako Cytomation commercial peroxidase blocking solution for 15 min, and the slides were washed again for 15 min in PBS. Unspecific binding of the antibodies was neutralized using Abcam protein block for 25 min. Excessive blocking solution was decanted prior to the incubation for 40 min with the primary antibody, and the Di-aminobenzidine/peroxidase reaction was followed under the microscope. The slides were washed in running tap water for 5 min and counterstained with Mayer’s Hematoxylin.
Slides evaluation
Photographs of four to five non-overlapping areas of the plated cells were taken using a 40× magnification, to provide a representative picture of the immunolabeling scenario. Given the cell homogeneity of the experimental system, the absolute fibroblasts count in one specific area was considered an objective denominator to normalize the number of peroxidase immunostained cells. Labeled percentage of total cells per 40× fields were averaged from 4-5 fields as described [20]. Furthermore, cell immunoreactivity was qualitatively graded using the scale of Galkowska et al. [21] considering the following score system: 0, no expression; 1-2, low; 3-4, mild; 5-6, high. The microphotographs were taken and scored by two researchers (JBA and YMM) unaware of the treatment group, through an artificial code introduced by an external investigator.
Statistical analysis
Data were processed using GraphPad Prism, version 6.0 for Windows (GraphPad Software, La Jolla California USA, www.graphpad.com). Results were expressed as mean ± standard deviation. Normal distribution (D’Agostino-Pearson omnibus) and variance homogeneity (Brown-Forsythe) tests were performed. Once demonstrated the normality of the data, comparisons between experimental groups were carried out using the two-tailed unpaired Student’s t test. In all cases, p-values lower than 0.05 were considered statistically significant.
RESULTS AND DISCUSSION
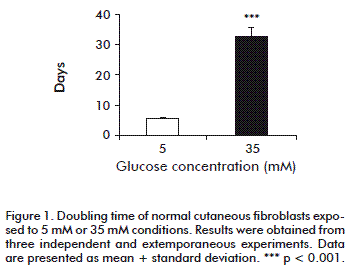
We calculated doubling time as an expression of the proliferation rate. Under the present experimental conditions, normal cutaneous fibroblasts exposed to 5 mM glucose exhibited a doubling time of 5.71 ± 0.55 days. However, the high-glucose environment increased doubling time to 32.15 ± 3.07 days (p < 0.0001), regardless the 15 % FBS supplementation (Figure 1). In contrast, the presence of 35 mM glucose did not affect proliferation (not shown), indicating that osmotic damage could be ruled out.
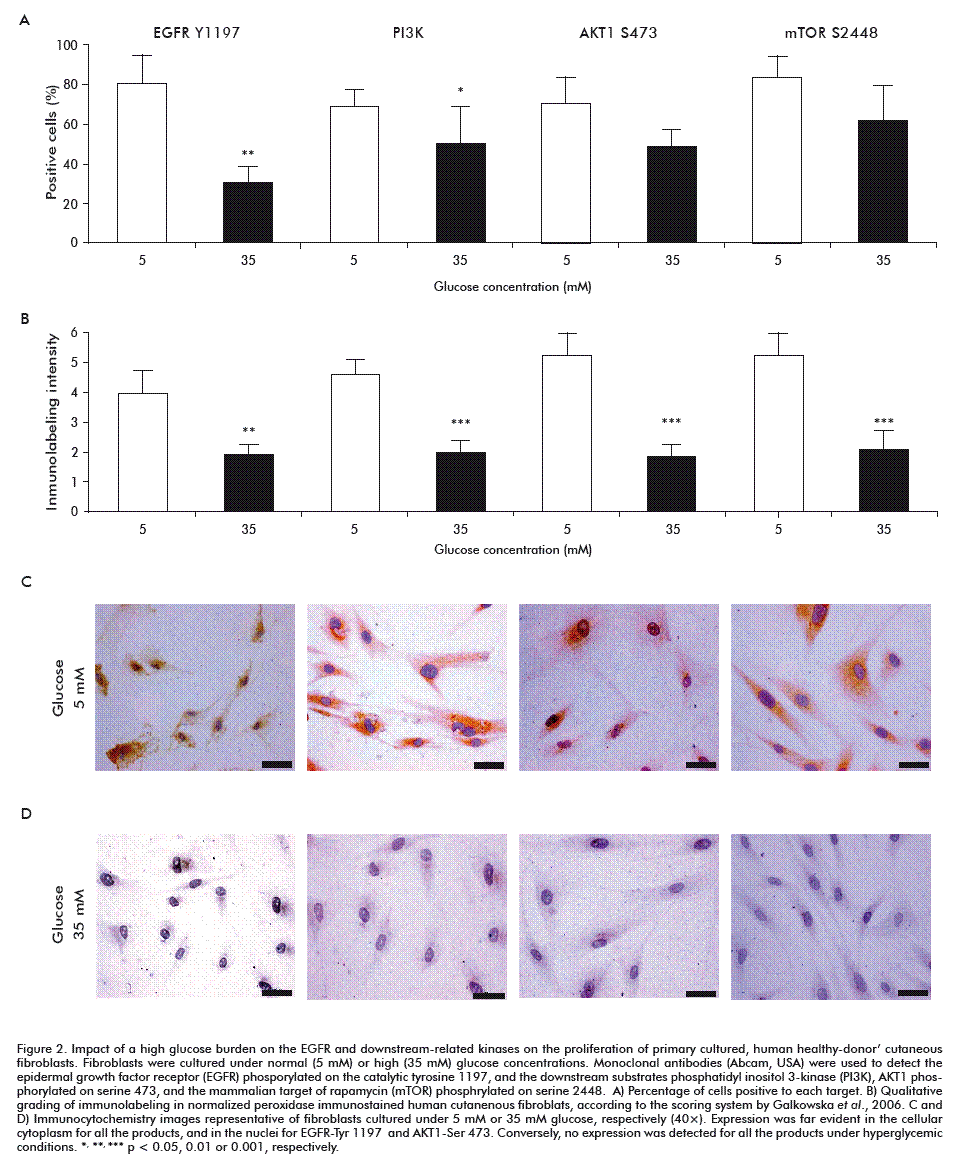
This proliferative delay appeared concomitant to a 50 % reduction in the percentage of EGFR positive cells phosphorylated on tyrosine residue 1197 (Figure 2), as compared to cells incubated under normal glucose concentrations (p = 0.004). The immunolabelling intensity was also significantly reduced (p < 0.0001) as a consequence of high glucose exposure. As demonstrated in prior studies, high glucose levels are toxic for healthy donor-cutaneous fibroblasts, by depressing critical pathways controlling cells’ survival, metabolism and proliferation. The novelty of this study lies in the demonstration that, in this type of cells, EGFR is likely a proximal target of high glucose concentrations, reducing the autophosphorylation of its catalytic tyrosine residue tyrosine-1197, which cause the attenuation of a signaling cascade that drives changes in gene expression, especially in cell proliferation [22].
In line with this, high glucose stress significantly reduced PI3K (p85 alpha subunit) expression in terms of percentage of positively labeled cells (p = 0.02) and the associated immunoreaction intensity (p < 0.001) as referred to fibroblasts grown under 5 mM glucose (Figure 2). This finding may be interpreted as resulting from the EGFR reduced autophosphorylation, since the PI3K p85 alpha subunit is an adaptor molecule that regulates the activity of the PI3K catalytic p110 subunit by binding to phosphorylated receptor tyrosine kinases (RTKs) through its SH2 domain and mediating the interaction between p110 and the plasma membrane [23].
Downstream, the master switch kinase AKT1 was also affected under a high glucose environment in relation to normoglucose-cultured fibroblasts. Although no statistical differences were detected (p = 0.11), there was a 20 % reduction in the percentage of cells positive to the active isoform which is phosphorylated on serine 473. However, the immunointensity reaction appeared significantly decreased to less than 50 % (p < 0.001; Figure 2). The serine/threonine protein kinase AKT1 is a major signal transducer of the PI3K pathway in all cells and tissues, and plays a pivotal role in the maintenance of cellular processes [24]. AKT1 is involved in controlling cell proliferation and survival, particularly by preventing apoptosis [25]. Although we found no statistical significance for the percentage of cells expressing phosphorylation on AKT1-serine 473 residue, it was quantitatively and qualitatively reduced in fibroblasts exposed to high glucose. This may stem from the upstream PI3K p85 expression level. AKT1 full activation requires two phosphatidylinositol-3,4,5-triphosphate-dependent phosphorylation events, while one of them relay on serine 473 [26, 27] for which the complex of the mammalian target of rapamycin (mTOR-Rictor complex) is critical [28].
We observed a similar behavior in the immunoreaction identifying one of the active isoforms of mTOR (specific phosphorylation on serine 2448). High glucose reduced the percentage of positive cells in about 23 %, with no statistical significance (p = 0.21). Moreover, the immunolabelling intensity score decreased to less than 50 % (p = 0.001) as compared to normal glucose environment fibroblasts (Figure 2). mTOR is a serine/threonine kinase which plays a key role as regulator of protein translation and sensor of nutrient status, thus controlling the balance between cell growth and autophagy [29-32]. The fact that signaling via mTOR and phosphorylation on serine 2448 is stimulated by growth factors and attenuated following amino acids starvation [33, 34], suggests the full disruption of this transduction axis from its proximal trigger; in this case, the EGFR. Conclusively, these findings highlight how intricate, yet finely regulated, the EGFR/PI3K/Akt/mTOR axis continuity is.
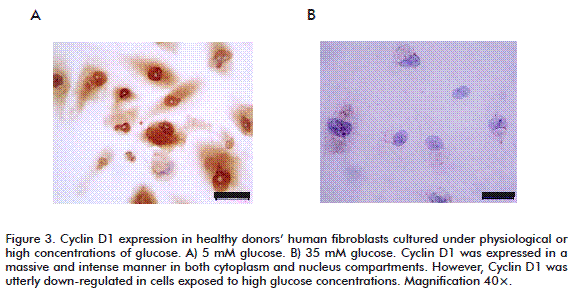
In addition, the high glucose-mediated cytotoxicity ultimately led to a complete inhibition of cyclin D1 expression (Figure 3A). This finding may theoretically explain the proliferative arrest detected in such cells. In contrast, under normal culture conditions, fibroblasts’ cyclin D1 was found intensively and massively expressed in both nuclear and cytoplasmic compartments (Figure 3B). Cyclin D1 responds to tyrosine kinase-activated receptors by acting as a growth factor sensor [35-37]. In chronic wounds, where growth factors are scarce or their titers are below a threshold level, signals from the wound environment are not able to activate the cell cycle machinery and no cyclin D1 is synthesized [20, 38].
It has been previously shown that high glucose exposure attenuates tyrosine-phosphorylation of EGFR in embryonic rat fibroblasts expressing human insulin receptors [39]. In that experimental system, the authors also detected an attenuation of PI3K kinase activity, with no significant MAPK kinase activity variation between normal and high glucose exposed cells. However, glucose burden did not affect proliferation rate, since DNA content remained the same in all the experimental conditions.
In our study, we detected a clear cut effect of glucose burden on EGFR phosphorylation status, specifically on Y1197 residue, and on PI3K expression levels. This significant reduction was accompanied with a 5-fold lengthening in doubling time, distinctive of a negative effect on the cell proliferation rate. Since our main goal was to understand diabetic wound healing failure from a molecular point of view, other key effectors were studied downstream in the EGFR signaling pathway. We additionally observed that the exposure to high glucose affected the expression of phosphorylated active intermediates, such as AKT1 (S473) and mTOR (s2448) and the key cell cycle pro-moter cyclin D1.
There was demonstrated in in vitro models that synthesis, proliferative and secreting capabilities are reduced in diabetics’ cutaneous fibroblasts [40-44]. High glucose concentration in the culture medium negatively impacted on the physiology of normal fibroblasts, thus inhibiting proliferation and turning the cells resistant to growth factors-stimulated mitogenesis [45-48]. Other groups reported that high glucose reduced collagen synthesis in a dose-dependent manner [12, 49]; and that cell migration speed is reduced by nearly 40 %, associated to a decrease in cell direc-tionality and to non-productive protrusive events due to cells’ polarization failure [50].
The mechanisms whereby high glucose concentrations can impair fibroblasts physiology, including proliferative capabilities, are not fully elucidated but had been associated to high L-lactate production [10, 44, 51] and the generation of cytotoxic reactive oxygen species [13, 14, 52]. We deem, however, that adducts formation between EGFR and AGEs precursors could be relevant to explain our findings. Previous experiments have proved significant AGEs accumulation in endothelial cells cultured for 7 days under glucose overload [53, 54] and that AGE modification of growth factors drastically reduced the cells mitogenic activity by 70 % [55, 56]. Obviously, this hypothesis requires intracellular AGEs concentration measurements and their chemical interaction with the EGFR in a time-window manner.
Taken together, our data show that high glucose concentration can hinder fibroblasts’ EGFR autophosphorylation and, consequently, deteriorate the activation of critical downstream pathways that may lead to fibroblasts proliferation delay and wound chronification. In a translational manner for clinical medicine, these findings emphasize on the need of a strict metabolic control of the diabetic patient affected by diabetic foot ulcers in order to facilitate the healing mechanism.
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
REFERENCES
1. International Diabetes Federation. Global Diabetes Plan 2011-2021. 2011 [cited 2014 Oct 9]. Available from: http://www.idf.org/sites/default/files/Global_Diabetes_Plan_Final.pdf.
2. Armstrong DG, Wrobel J, Robbins JM. Guest Editorial: are diabetes-related wounds and amputations worse than cancer? Int Wound J. 2007;4(4):286-7.
3. Monteiro-Soares M, Martins-Mendes D, Vaz-Carneiro A, Dinis-Ribeiro M. Lower limb amputation following foot ulcers in patients with diabetes: classification systems’ external validation and comparative analysis. Diabetes Metab Res Rev. 2014. DOI: 10.1002/dmrr.2634.
4. Jupiter DC, Thorud JC, Buckley CJ, Shibuya N. The impact of foot ulceration and amputation on mortality in diabetic patients. I: From ulceration to death, a systematic review. Int Wound J. 2015. DOI: 10.1111/iwj.12404.
5. Pickwell K, Siersma V, Kars M, Apelqvist J, Bakker K, Edmonds M, et al. Predictors of lower-extremity amputation in patients with an infected diabetic foot ulcer. Diabetes Care. 2015;38(5):852-7.
6. Zhan LX, Branco BC, Armstrong DG, Mills JL, Sr. The Society for Vascular Surgery lower extremity threatened limb classification system based on Wound, Ischemia, and foot Infection (WIfI) correlates with risk of major amputation and time to wound healing. J Vasc Surg. 2015;61(4):939-4.
7. Armstrong DG, Cohen K, Courric S, Bharara M, Marston W. Diabetic foot ulcers and vascular insufficiency: our population has changed, but our methods have not. J Diabetes Sci Technol. 2011;5(6):1591-5.
8. Berlanga-Acosta J, Valdés-Pérez C, Savigne-Gutierrez W, Mendoza Marí Y, Franco-Pérez N, Varga-Marchiran E, et al. Cellular and molecular insights into the wound healing mechanism in diabetes. Biotecnol Apl. 2010;27(4):255-61.
9. Berlanga-Acosta J, Schultz GS, Lopez-Mola E, Guillen-Nieto G, Garcia-Siverio M, Herrera-Martinez L. Glucose toxic effects on granulation tissue productive cells: the diabetics’ impaired healing. Biomed Res Int. 2013;2013:256043.
10. Hehenberger K, Heilborn JD, Brismar K, Hansson A. Inhibited proliferation of fibroblasts derived from chronic diabetic wounds and normal dermal fibroblasts treated with high glucose is associated with increased formation of l-lactate. Wound Repair Regen. 1998;6(2):135-41.
11. Yevdokimova NY. High glucose-induced alterations of extracellular matrix of human skin fibroblasts are not dependent on TSP-1-TGFbeta1 pathway. J Diabetes Complications. 2003;17(6):355-64.
12. Andreea SI, Marieta C, Anca D. AGEs and glucose levels modulate type I and III procollagen mRNA synthesis in dermal fibroblasts cells culture. Exp Diabetes Res. 2008;2008:473603.
13. Lamers ML, Almeida ME, Vicente-Manzanares M, Horwitz AF, Santos MF. High glucose-mediated oxidative stress impairs cell migration. PLoS One. 2011;6(8):e22865.
14. Yu P, Wang Z, Sun X, Chen X, Zeng S, Chen L, et al. Hydrogen-rich medium protects human skin fibroblasts from high glucose or mannitol induced oxidative damage. Biochem Biophys Res Commun. 2011;409(2):350-5.
15. Xuan YH, Huang BB, Tian HS, Chi LS, Duan YM, Wang X, et al. High-glucose inhibits human fibroblast cell migration in wound healing via repression of bFGF-regulating JNK phosphorylation. PLoS One. 2014;9(9):e108182.
16. Kusewitt DF, Choi C, Newkirk KM, Leroy P, Li Y, Chavez MG, et al. Slug/Snai2 is a downstream mediator of epidermal growth factor receptor-stimulated reepithelialization. J Invest Dermatol. 2009;129(2):491-5.
17. Portero-Otin M, Pamplona R, Bellmunt MJ, Ruiz MC, Prat J, Salvayre R, et al. Advanced glycation end product precursors impair epidermal growth factor receptor signaling. Diabetes. 2002;51(5):1535-42.
18. Doronzo G, Viretto M, Russo I, Mattiello L, Anfossi G, Trovati M. Effects of high glucose on vascular endothelial growth factor synthesis and secretion in aortic vascular smooth muscle cells from obese and lean zucker rats. Int J Mol Sci. 2012;13(8):9478-88.
19. Roth V. Doubling Time. 2006 [cited 2014 Oct 9]. Available from: http://www.doubling-time.com/compute.php.
20. Vande Berg JS, Rose MA, Payne WG, Haywood-Reid PL, Robson MC. Significance of cell cycle for wound stratification in clinical trials: analysis of a pressure ulcer clinical trial utilizing cyclin D/cdk4. Wound Repair Regen. 2003;11(1):11-8.
21. Galkowska H, Wojewodzka U, Olszewski WL. Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen. 2006;14(5):558-65.
22. Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19(13):3159-67.
23. Arcaro A, Guerreiro AS. The phosphoinositide 3-kinase pathway in human cancer: genetic alterations and therapeutic implications. Curr Genomics. 2007;8(5):271-306.
24. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261-74.
25. Chin YR, Toker A. Function of Akt/ PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009;21(4):470-6.
26. Partovian C, Simons M. Regulation of protein kinase B/Akt activity and Ser473 phosphorylation by protein kinase Calpha in endothelial cells. Cell Signal. 2004;16(8):951-7.
27. Zhuo DX, Zhang XW, Jin B, Zhang Z, Xie BS, Wu CL, et al. CSTP1, a novel protein phosphatase, blocks cell cycle, promotes cell apoptosis, and suppresses tumor growth of bladder cancer by directly dephosphorylating Akt at Ser473 site. PloS One. 2013;8(6):e65679.
28. Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280(49):40406-16.
29. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163-75.
30. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098-101.
31. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8(3):224-36.
32. Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323-8.
33. Rosner M, Siegel N, Valli A, Fuchs C, Hengstschlager M. mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids. 2010;38(1):223-8.
34. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40(2):310-22.
35. Moriuchi A, Hirono S, Ido A, Ochiai T, Nakama T, Uto H, et al. Additive and inhibitory effects of simultaneous treatment with growth factors on DNA synthesis through MAPK pathway and G1 cyclins in rat hepatocytes. Biochem Biophys Res Commun. 2001;280(1):368-73.
36. Cao C, Huang X, Han Y, Wan Y, Birnbaumer L, Feng GS, et al. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009;2(68):ra17.
37. Alam S, Pal A, Kumar R, Dwivedi PD, Das M, Ansari KM. EGFR-mediated Akt and MAPKs signal pathways play a crucial role in patulin-induced cell proliferation in primary murine keratinocytes via modulation of Cyclin D1 and COX-2 expression. Mol Carcinog. 2014;53(12):988-98.
38. Lee do H, Choi KH, Cho JW, Kim SY, Kwon TR, Choi SY, et al. Recombinant growth factor mixtures induce cell cycle progression and the upregulation of type I collagen in human skin fibroblasts, resulting in the acceleration of wound healing processes. Int J Mol Med. 2014;33(5):1147-52.
39. Obata T, Maegawa H, Kashiwagi A, Pillay TS, Kikkawa R. High glucose-induced abnormal epidermal growth factor signaling. J Biochem. 1998;123(5):813-20.
40. Rowe DW, Starman BJ, Fujimoto WY, Williams RH. Abnormalities in proliferation and protein synthesis in skin fibroblast cultures from patients with diabetes mellitus. Diabetes. 1977;26(4):284-90.
41. Loots MA, Lamme EN, Mekkes JR, Bos JD, Middelkoop E. Cultured fibroblasts from chronic diabetic wounds on the lower extremity (non-insulin-dependent diabetes mellitus) show disturbed proliferation. Arch Dermatol Res. 1999;291(2-3):93-9.
42. Lerman OZ, Galiano RD, Armour M, Levine JP, Gurtner GC. Cellular dysfunction in the diabetic fibroblast: impairment in migration, vascular endothelial growth factor produc¬tion, and response to hypoxia. Am J Pathol. 2003;162(1):303-12.
43. Lobmann R, Pap T, Ambrosch A, Waldmann K, Konig W, Lehnert H. Differential effects of PDGF-BB on matrix metalloproteases and cytokine release in fibroblasts of Type 2 diabetic patients and normal controls in vitro. J Diabetes Complications. 2006;20(2):105-12.
44. Moruzzi N, Del Sole M, Fato R, Gerdes JM, Berggren PO, Bergamini C, et al. Short and prolonged exposure to hyperglycaemia in human fibroblasts and endothelial cells: metabolic and osmotic effects. Int J Biochem Cell Biol. 2014;53:66-76.
45. Sibbitt WL, Jr., Mills RG, Bigler CF, Eaton RP, Griffey RH, Vander Jagt DL. Glucose inhibition of human fibroblast proliferation and response to growth factors is prevented by inhibitors of aldose reductase. Mech Ageing Dev. 1989;47(3):265-79.
46. Hehenberger K, Hansson A. High glucose-induced growth factor resistance in human fibroblasts can be reversed by antioxidants and protein kinase C-inhibitors. Cell Biochem Funct. 1997;15(3):197-201.
47. Cubbon RM, Ali N, Sengupta A, Kearney MT. Insulin- and growth factor-resistance impairs vascular regeneration in diabetes mellitus. Curr Vasc Pharmacol. 2012;10(3):271-84.
48. Tecilazich F, Dinh TL, Veves A. Emerging drugs for the treatment of diabetic ulcers. Expert Opin Emerg Drugs. 2013;18(2):207-17.
49. Trevisan R, Yip J, Sarika L, Li LK, Viberti G. Enhanced collagen synthesis in cultured skin fibroblasts from insulin-dependent diabetic patients with nephropathy. J Am Soc Nephrol. 1997;8(7):1133-9.
50. Grazul-Bilska AT, Luthra G, Reynolds LP, Bilski JJ, Johnson ML, Adbullah SA, et al. Effects of basic fibroblast growth factor (FGF-2) on proliferation of human skin fibroblasts in type II diabetes mellitus. Exp Clin Endocrinol Diabetes. 2002;110(4):176-81.
51. Hehenberger K, Hansson A, Heilborn JD, Abdel-Halim SM, Ostensson CG, Brismar K. Impaired proliferation and increased L-lactate production of dermal fibroblasts in the GK-rat, a spontaneous model of non-insulin dependent diabetes mellitus. Wound Repair Regen. 1999;7(1):65-71.
52. Tsai CH, Chiang YC, Chen HT, Huang PH, Hsu HC, Tang CH. High glucose induces vascular endothelial growth factor production in human synovial fibroblasts through reactive oxygen species generation. Biochim Biophys Acta. 2013;1830(3):2649-58.
53. McPherson JD, Shilton BH, Walton DJ. Role of fructose in glycation and cross-linking of proteins. Biochemistry. 1988;27(6):1901-7.
54. Queisser MA, Yao D, Geisler S, Hammes HP, Lochnit G, Schleicher ED, et al. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010;59(3):670-8.
55. Giardino I, Edelstein D, Brownlee M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J Clin Invest. 1994;94(1):110-7.
56. Liu H, Yu S, Zhang H, Xu J. Angiogenesis impairment in diabetes: role of methylglyoxal-induced receptor for advanced glycation endproducts, autophagy and vascular endothelial growth factor receptor 2. PloS One. 2012;7(10):e46720.
Received in October 2014.
Accepted in December 2014.
Jorge Berlanga-Acosta. Tissue Repair and Cyto-protection Laboratory, Direction of Biomedical Research. Center for Genetic Engineering and Biotechnology, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba. E-mail: jorge.berlanga@cigb.edu.cu.


{kind=link}
{kind=link}