










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.32 no.2 La Habana Apr.-June 2015
RESEARCH
Parenteral delivery of the vaccine candidate TERAVAC-HIV-1 bypasses pre-existing immune response to the hepatitis B virus antigens in mice
La administración parenteral del candidato vacunal TERAVAC-HIV-1 no es interferida por la respuesta inmune previa contra antígenos del virus de la hepatitis B en ratones
Enrique Iglesias, Daymir García, Julio C Aguilar
Departamento de Vacunas, Dirección de Investigaciones Biomédicas, Centro de Ingeniería Genética y Biotecnología. Ave. 31 e/158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.
ABSTRACT
The recombinant hepatitis B virus surface (HBsAg) and core (HBcAg) virus like particles (VLPs) have the ability to serve as carriers of foreign B cell and CTL epitopes. Different approaches have been used to couple the target epitopes, like the insertion into the primary sequence of the VLP, covalent and noncovalent linkage. Particularly, the non-covalent linkage was used to develop the vaccine formulation Teravac against the human immunodeficiency virus type 1 (HIV-1). Teravac is an aggregate of the recombinant protein CR3 of HIV-1 and both HBV VLPs. Previous studies have shown that immunization of Teravac in mice induced a Th1 response with CD8+ T cells. However, because millions of people are infected with the HBV and millions of doses of the HBV vaccine have been administered worldwide, the pre-existing immune response to the HBcAg and/or HBsAg is a rather frequent event. This opens de question about the impact of the anti-HBc and/or anti-HBs antibody response on the CR3(HIV)-specific cellular response elicited with Teravac. To answer this question, the effect of the pre-existing anti-HBc and the combined anti-HBc/anti-HBs antibodies was studied in mice. Our findings suggest that the induction of CR3(HIV)-specific cellular responses of CD4+ and CD8+ cells are not impaired by pre-existing high IgG titers in either situation.
Keywords: HIV vaccines, cellular immune response, hepatitis B antibodies, hepatitis B core antigens, hepatitis B surface antigen.
RESUMEN
Los antígenos recombinantes de superficie y de la cápsida del virus de la hepatitis B (HBsAg y HBcAg, respectivamente) estructurados como partículas similares a virus (VLP) sirven como portadores de epitopos de células B y linfocitos T citotóxicos heterólogos. Tales epitopos se acoplan mediante estrategias como la inserción en la secuencia primaria de las VLP, o mediante unión covalente o no covalente. Esta última se usó para desarrollar la formulación vacunal Teravac contra el virus de la inmunodeficiencia humana tipo 1 (VIH-1). Teravac es un agregado entre la proteína CR3 derivada del VIH-1 y las VLP de ambos antígenos del HBV. Estudios previos mostraron que la inmunización con Teravac en ratones indujo una respuesta de células T CD8+ de tipo Th1. Sin embargo, millones de personas son infectadas con el HBV y se han administrado millones de dosis vacunales contra este virus, con la frecuente inducción de respuesta inmune contra el HBcAg, el HBsAg o ambos. Con vistas a indagar sobre el posible impacto de la respuesta de anticuerpos anti-HBc, anti-HBs o ambas sobre la respuesta celular específica generada contra el antígeno CR3 al inmunizar con Teravac, se estudió el efecto de la respuesta pre-existente de anticuerpos anti-HBc y de su combinación con anticuerpos anti-HBs sobre la respuesta de linfocitos CD4+ y CD8+ en ratones. Nuestros resultados sugieren que la inducción de dicha respuesta celular contra el antígeno CR3 del VIH-1 no es interferida por los altos títulos de anticuerpos de subclase IgG generados con cualquiera de las variantes de VLP ensayadas.
Palabras clave: vacunas contra VIH, respuesta inmune celular, anticuerpos contra hepatitis B, antígeno de la cápsida de la hepatitis B, antígeno de superficie de la hepatitis B.
INTRODUCTION
Success in controlling the viral load, and an impressive improvement in the quality of life and life expectancy, have been achieved with antiretroviral therapies in HIV+ patients [1]. But, in spite of the hypothetical impact on the prevention of transmission expected with the 90-90-90 strategy proposed by UNAIDS [2] and the prospect of success with the PreP, a huge number of limitations in the field have to be surmounted. Regarding to this, it is known that low and lower middle-income countries lack financial support and sanitary infrastructure to achieve long-term implementation of these programs [3]. That is why many scientists still consider vaccination as the best measure to control the pandemic [4, 5].
In line with the vaccination strategy, a multiantigenic vaccine candidate named Teravac was developed, to induce essentially an anti-HIV-1 cellular immune response. This formulation contains aggregates of the recombinant protein CR3 with the surface (HBsAg, S) and core (HBcAg, C) virus-like particles (VLPs) of the hepatitis B virus (HBV)[6, 7]. The CR3 protein is a subunit antigen comprising several T helper (Th) and cytotoxic T lymphocyte (CTL) rich regions of HIV-1 proteins. The HBV antigens allow an effective adjuvant effect through nasal and subcutaneous immunizations, Th1 bias of the CR3(HIV-1)- specific immune response, and the induction of CD8+ T cells in the spleen and IFN-g-secreting cells in mesenteric lymph nodes [6, 8].
In the past, a pre-existing immune response to the carrier protein or immunity to the viral vector hindered vaccine candidates from eliciting effective immune responses [9, 10]. In the case of Teravac, it is important to consider that an effective vaccine against the HBV (based on the HBsAg) has been in place for more than 20 years and vaccination has integrated into the infant vaccination programs in several countries [11]. Moreover, HBV vaccination is recommended for HIV infected patients. Hence, a pre-existing immune response to the HBsAg becomes a highly probable scenario when administering a vaccine against HIV-1. Similarly, the presence of HBcAg-specific antibodies (anti-HBc) is a hallmark of chronic hepatitis B and its persistent carrier state. It has been estimated that approximately 248 million people were chronically infected with HBV worldwide in 2010 [12]. Thus, pre-existing anti-HBc IgG antibodies is also a common scenario. Additionally, around 88-95 % of adults spontaneously recover from HBV infection and develop anti-HBs (HBsAg-specific) and anti-HBc IgG antibodies [13].
One possible drawback of using HBV VLPs (HBcAg and HBsAg) in Teravac would be the suppression of their adjuvant effect on the CR3(HIV)-specific immune response by pre-existing VLP-specific antibodies. Fortunately, previous studies have evidenced that pre-existing anti-HBc antibodies have a marginal effect on the cellular and humoral response against heterologous fused antigens [14- 16]. Other authors have published similar results for the HBsAg [17]. Previously, we reported a significant Th1 adjuvant effect of both HBV VLPs on the CR3(HIV-1)-specific immune response as part of the multiantigenic formulation Teravac after subcutaneous immunization [6]. However, the effect of the HBcAg and HBsAg-specific pre-existing immune response was not studied. In the present study, the capacity of Teravac to stimulate the proliferation of CR3(HIV)-specific CD4+ and CD8+ T cells in the presence of an ongoing anti-HBc and anti-HBc/anti- HBs immune responses was explored.
MATERIALS AND METHODS
Antigens
The entire recombinant (r)HBcAg particle of 183 amino acids was expressed in Escherichia coli and the rHBsAg subtype adw2 in the yeast Pichia pastoris. The purification processes for both antigens were published [18, 19].
The HIV-1 antigen CR3 is composed of cytotoxic T lymphocyte (CTL) and helper T cell (Th) epitope-rich regions comprising T1 (Env421-440, protein location in HXB2 isolate), T2 (Env105-117) and the V3 loop (Env305- 318) from gp120, an epitope from gp41 (Env584-594), another from Vpr (Vpr66-80), a fragment of the p66/p51 (reverse transcriptase; RT) protein (Pol191-347), a part of Nef (Nef43-150), and a part of p24 Gag (Gag219-307). It was purified from E. coli BL21-CodonPlus(DE3)-RIL (Stratagene, La Jolla, CA) and the purification process was as described previously [6].
All antigens were pyrogen-free products with more than 95% purity [6, 18, 19].
Immunizations
Four groups of 6-8-weeks-old female Balb/c mice, purchased from CENPALAB (Havana, Cuba), were immunized in two rounds as shown in figure 1. Preimmune sera were obtained two days ahead of the immunizations. In the first phase, 12 animals per group were inoculated twice on days 0 and 13 with: groups 1 and 2, phosphate-buffered saline (PBS; placebo); 3, HBcAg (C) and 4, mixture of HBcAg and HBsAg (C+S). In the second phase, the animals received three additional inoculations on days 35, 56, 77 with: group 1, Teravac (positive control); 2, C+S (negative control); 3 and 4, Teravac (experimental groups). In all cases immunogens were prepared a day before and stored at 4°C until inoculation. They were administered subcutaneously in a 100 μL volume, adjuvanted in 1 mg/mL aluminum hydroxide (AlOOH; Superfos Biosector A/S, Vedbaek, Denmark). The dose for all antigens was 5 mg/mouse. The experiment and care of animals was conducted in accordance with institutional guidelines to avoid unnecessary suffering.
Serology
To assess the induction of anti-HBc and anti-HBs IgG-specific antibodies at the end of the phase 1, on day 34 animals’ sera were tested by an indirect EIA as reported [8]. Briefly, high binding capacity 96-well plates (Corning Life Sciences, Acton, MA) were coated with the antigen at 5 μg/mL. Plates were blocked with 2 % skim milk in PBS for 1 h at 37 °C. Subsequently, they were incubated with serum samples diluted in 1 % skim milk, and 1 % Tween 20 in PBS for 2 h at 37 °C. Rabbit anti-mouse total IgG peroxidase conjugate (MP Biomedicals, Aurora, OH) was incubated for 1 h at 37 °C. The reactions were then developed with substrate solution (52 mM Na2HPO4, 25 mM sodium citrate, 1 mg/mL o-phenylenediamine, and 0.1 % H2O2) for 10 min at room temperature (21 °C). The reaction was stopped with 3 M H2SO4. Last, absorbance was measured at 492 nm (A492nm) in a microplate reader (MultiskanFC, Finland). Five washes with 0.05 % Tween 20 in distilled water were carried out between each step.
Sera titers were calculated as the antilog of the resulting value after interpolation of the decimal logarithm of the absorbance values at a fixed serum dilution into a log-log linear regression analysis plotting dilution versus A492nm of the standard curve of a known titer serum. The titer of the standard curve was defined as the highest dilution that gave more than twice the absorbance of the negative control serum diluted 1:100. Total IgG titers were expressed in Standard Units (STD Units) and reported as the geometric mean plus 95 % confidence interval (CI) of the individual sera.
Proliferation of CD4+ and CD8+ cells
The proliferation of CD4+ and CD8+ cells from splenocytes of five randomly selected mice per group was assessed using CFSE staining and ex vivo stimulation with the CR3 antigen as reported [8]. Then, 1×106 cells were selectively stained with 0.2 mg of anti-CD4-APC (clone L3T4) or anti-CD8a-APC (clone Ly-2) (Biosciences, San Diego, CA, USA), respectively. They were washed and analyzed for the expression of the surface marker. Samples were acquired using a PAS III flow cytometer (Partec GmbH, Münster, Germany) and the analysis was performed with Flomax v2.4f software (Partec GmbH). Ten thousand viable lymphocytes including blasts (gate R1) were gated for the analysis. The frequency of CR3-specific CD4+ and CD8+ T cells that divided after ex vivo stimulation was determined by subtracting the percentage of CFSElow cells from unstimulated cultures from the percentage of CFSElow splenocytes stimulated with the Ag. Data from proliferation studies were displayed as the mean percentage of CD4+/8+ CFSElow cells plus 95 % CI from five individual mice per group.
Statistical analyses
Statistical analysis was carried out using GraphPad Prism version 5 Software (GraphPad Software, San Diego, CA, USA). Unpaired Student’s t test with Welch’s correction and Kruskal-Wallis with Dunn’s multiple comparisons test vs control group were employed to assess significant differences. A p value lower than 0.05 was considered statistically significant.
RESULTS AND DISCUSSION
In this study, we were interested in to evaluate the impact of the pre-existing immune response to the surface and core antigens of HBV on the cellular immune response elicited after parenteral inoculation of Teravac. Two different scenarios were evaluated. First, Balb/c mice were inoculated with the HBcAg in alum to simulate the condition of chronic carriers of the HBV who are characterized by the presence of HBcAg-specific IgG antibodies. Second, animals were inoculated with a mixture of HBcAg and HBsAg to elicit antibodies against both antigens. This has been observed in persons with natural immunity to the HBV or those with previous exposure and recovery from the infection with loss of detectable anti-HBs that develop detectable anti-HBs after vaccination [13]. The pre-existing of anti-HBc and anti-HBs antibodies represents the worst case scenario.
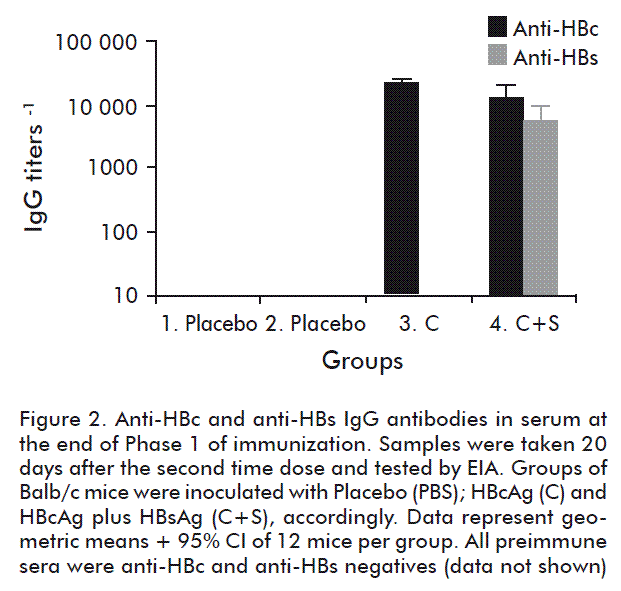
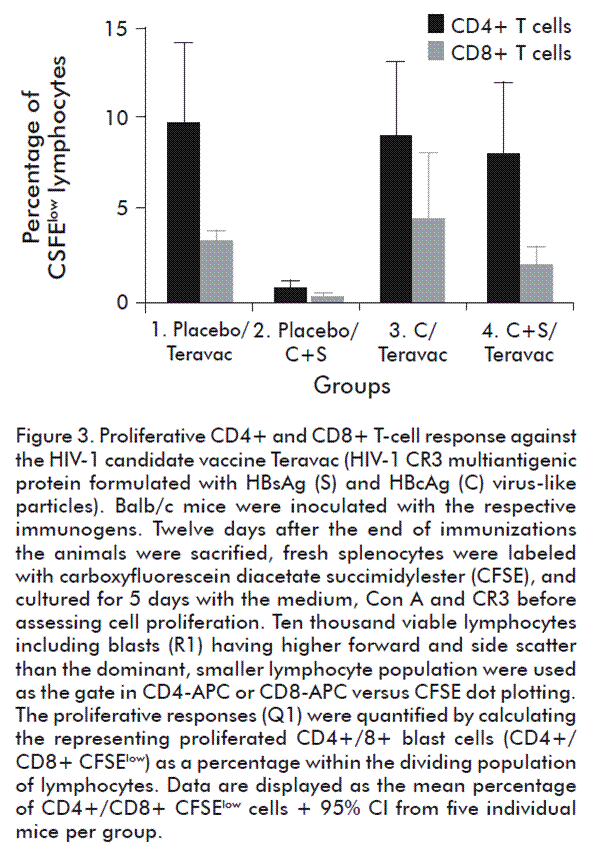
At the end of the first phase of immunization, all the animals in groups 3 (HBcAg (C)) and 4 (HBcAg and HBsAg) seroconverted to HBcAg and HBsAg. As shown in figure 2, it resulted in the generation of similar high levels of anti-HBc IgG antibodies in both groups (p > 0.05, unpaired Student’s t test with Welch’s correction). Also, an important level of anti-HBs IgG antibodies was elicited in the group 4. In consequence, a pre-existing antibody response was generated in the animals. Then, in the second phase of immunization animals were further inoculated three times with Teravac, except the group 2 that was inoculated with the mixture of HBcAg and HBsAg as a negative control. Twelve days later, the proliferation of CR3(HIV)-specific CD4+ and CD8+ cells was assessed by flow cytometry. As shown in figure 3, proliferation of CD4+ cells was not abrogated by the presence of high anti-HBcAg IgG titers (see group 3) or the combination of anti-HBcAg and anti-HBsAg IgG antibodies (group 4). In fact, similar levels of proliferation was verified in both experimental groups when compared with the positive control group 1 (p > 0.05; Kruskal-Wallis and Dunn’s multiple comparisons test vs control group). When analyzing the proliferation of CD8+ cells, we noted a slightly lower average response only in group 4 in which a pre-existing anti-HBc and anti-HBs immune responses were elicited. Nevertheless, no statistically significant differences in these values could be documented when compared with the positive control group 1 (p > 0.05; Kruskal-Wallis and Dunn’s multiple comparisons test vs control group).
Our findings suggest that the CR3(HIV)-specific cellular responses of CD4+ and CD8+ cells induced by subcutaneous immunization with Teravac are not impaired by the pre-existing immune response to a single or both major structural HBV antigens. To our knowledge, this is the first report evaluating the effect of the pre-existing immune response for both VLPs of HBV on the immune response against a co-administered antigen. We did not test the effect of a pre-existing immune response to the HBsAg alone to assess the situation of vaccinated people without previous exposure to HBV. But, considering our findings together with those of Netter et al. [17], it suggests that such pre-existing response will not interfere in the effect attained by immunizing with Teravac.
Nevertheless, the current experimental setting has several limitations. First, the pre-existing immunity for HBV was simulated by vaccination with antigens since the mouse model does not become infected by HBV. Second, we inoculated recombinant antigens in adjuvant instead of using natural antigens recovered from patients or a whole virus lysate. Third, the pre-existing immunity was simulated for only two antigens of the HBV, not ruling out the influence of some immunopathological events due to other HBV antigens in humans. Fourth, we did not simulate the influence of immunopathological events associated with the HBV/ HIV-1 coinfection to address the condition of HBV+/ HIV-1+ patients. Hence, the experimental conditions did not reproduce neither the immunopathological events during chronic HBV infection in humans nor the condition of HBV/HIV-1-coinfected patients.
But, it is known that the immune response to the HBsAg and HBcAg in mice resembles the human response [20]. In fact, the HBsAg is the active pharmaceutical ingredient of prophylactic vaccines currently in use against HBV, and the in vivo potency test for the release of vaccine batches is assessed in mice because the HBsAg-specific IgG antibody response elicited in this species correlates well with the protective response in humans [21]. Therefore, despite the fact that the model used in this research did not rule out the influence of immunopathological events related to the HBV infection, it was still relevant to assess the impact of the anti-HBc and anti-HBs/anti-HBc IgG response on the CR3(HIV-1)-specific T cell response. Moreover, it could be also relevant in the case of HBV+/HIV-1+ patients. In these patients, when the nadir of CD4+ T cell counts is higher than 350 cells/ μL and the HIV viral load is undetectable or very low (as under antiretroviral treatment), functional immunocompetence is preserved [22]. Thus, the findings in the mouse model are still useful to predict possible outcomes in humans vaccinated with Teravac.
The possibility to induce an anti-HIV-1 cellular response after immunization with Teravac in the presence of pre-existing immune responses to the HBV could be considered an advantage of this vaccine candidate in the therapeutic setting. In fact, there has been estimated that around 2-4 millions of HIV-infected individuals worldwide are also co-infected with HBV [23]. Moreover, it is important to consider that it is in the Subsaharan Africa where the prevalence of HBV is the highest [12] as well as for HIV-1 [23]. Taking into account the high prevalence of HBV-infected and HIV-1/HBV-coinfected patients in some geographical areas, it is important to notice that a new vaccine formulation called Nasvac is under development by our group for the treatment of chronic HBV infection, which successfully combines both the HBsAg and HBcAg antigens [24, 25]. Because the vaccine candidate Teravac comprises the same previous two antigens of the HBV plus CR3 from HIV-1, we speculate that HBV-infected and HIV-1/HBV-coinfected patients might also benefit from Teravac vaccination to achieve some control over the HBV viral load. That is important since HIV-1/HBV-coinfection is associated with lower T CD4+ counts [26].
The potential negative impact of the pre-existing anti-HBs, anti-HBc or the combined anti-HBcAg/ anti-HBs IgG response on the HIV-1-specific cellular immune response elicited after inoculation with the vaccine candidate Teravac should be investigated in humans. This is paramount for HIV– individuals (prophylactic scenario) and HIV+ patients (therapeutic scenario) vaccinated against the HBV, chronically infected, convalescents or recovered from HBV infection.
CONCLUSIONS
The findings described in this study revealed no evidence that induction of CR3(HIV)-specific cellular responses of CD4+ and CD8+ cells after immunization with Teravac are impaired by pre-existing immune response to a single or both major structural antigens of HBV in mice.
ACKNOWLEDGEMENTS
This work was supported by the Center for Genetic Engineering and Biotechnology, Havana, Cuba. There are no competing financial interests. We thank Ismariley Revé, Sara Clark and Lariza Gorobaya for their technical assistance.
REFERENCES
1. Samji H, Cescon A, Hogg RS, Modur SP, Althoff KN, Buchacz K, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One. 2013;8(12):e81355.
2. UNAIDS. Fast track ending the AIDS epidemic by 2030. Geneva: UNAIDS; 2014 [cited 2015 Nov 14]. Available from: www.unaids.org/sites/default/files/media_asset/JC2686_WAD2014report_en.pdf
3. Grimsrud A, Balkan S, Casas EC, Lujan J, Van Cutsem G, Poulet E, et al. Outcomes of antiretroviral therapy over a 10-year period of expansion: a multicohort analysis of African and Asian HIV programs. J Acquir Immune Defic Syndr. 2014;67(2):e55-66.
4. Chawla S, Sahoo SS, Jain R, Khanna P, Mehta B, Singh I. HIV: is a vaccine the answer? Hum Vaccin Immunother. 2014;10(1):238-40.
5. Fauci AS, Marston HD. Ending AIDS--is an HIV vaccine necessary? N Engl J Med. 2014;370(6):495-8.
6. Iglesias E, Thompson R, Carrazana Y, Lobaina Y, Garcia D, Sanchez J, et al. Coinoculation with hepatitis B surface and core antigen promotes a Th1 immune response to a multiepitopic protein of HIV- 1. Immunol Cell Biol. 2006;84(2):174-83.
7. Guillen G, Aguilar JC, Dueñas S, Hermida L, Iglesias E, Penton E, et al. Virus-like particles as nanovaccine candidates. Adv Nat Sci Nanosci Nanotechnol. 2013;4(1):015005.
8. Iglesias E, Garcia D, Carrazana Y, Aguilar JC, Sanchez A, Gorobaya L, et al. Anti-HIV-1 and anti-HBV immune responses in mice after parenteral and nasal co-administration of a multiantigenic formulation. Curr HIV Res. 2008;6(5):452-60.
9. Schutze MP, Leclerc C, Jolivet M, Audibert F, Chedid L. Carrier-induced epitopic suppression, a major issue for future synthetic vaccines. J Immunol. 1985;135(4):2319-22.
10. Fausther-Bovendo H, Kobinger GP. Pre-existing immunity against Ad vectors: humoral, cellular, and innate response, what’s important? Hum Vaccin Immunother. 2014;10(10):2875-84.
11. Meireles LC, Marinho RT, Van Damme P. Three decades of hepatitis B control with vaccination. World J Hepatol. 2015;7(18):2127-32.
12. Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet. 2015;386(10003):1546-55.
13. WHO. Hepatitis B: WHO/CDS/CSR/LYO/2002.2. Department of Communicable Diseases Surveillance and Response. 2002 [cited 2015 Nov 11]. Available from: http://www.who.int/emc.
14. Schwarz K, Meijerink E, Speiser DE, Tissot AC, Cielens I, Renhof R, et al. Efficient homologous prime-boost strategies for T cell vaccination based on virus-like particles. Eur J Immunol. 2005;35(3):816-21.
15. Ruedl C, Schwarz K, Jegerlehner A, Storni T, Manolova V, Bachmann MF. Virus-like particles as carriers for T-cell epitopes: limited inhibition of T-cell priming by carrier-specific antibodies. J Virol. 2005;79(2):717-24.
16. Schodel F, Peterson D, Milich D. Hepatitis B virus core and e antigen: immune recognition and use as a vaccine carrier moiety. Intervirology. 1996;39(1-2):104-10.
17. Netter HJ, Woo WP, Tindle R, Macfarlan RI, Gowans EJ. Immunogenicity of recombinant HBsAg/HCV particles in mice pre-immunised with hepatitis B virus-specific vaccine. Vaccine. 2003;21(21-22):2692-7.
18. Hardy E, Martinez E, Diago D, Diaz R, Gonzalez D, Herrera L. Large-scale production of recombinant hepatitis B surface antigen from Pichia pastoris. J Biotechnol. 2000;77(2-3):157-67.
19. Lobaina Y, Garcia D, Abreu N, Muzio V, Aguilar JC. Mucosal immunogenicity of the hepatitis B core antigen. Biochem Biophys Res Commun. 2003;300(3):745-50.
20. Milich DR. Immune response to hepatitis B virus proteins: relevance of the murine model. Semin Liver Dis. 1991;11(2):93-112.
21. WHO. Expert Committee on Biological Standardization, sixty-first report. Annex 4. Geneva: WHO; 2013.
22. Lange CG, Lederman MM, Medvik K, Asaad R, Wild M, Kalayjian R, et al. Nadir CD4+ T-cell count and numbers of CD28+ CD4+ T-cells predict functional responses to immunizations in chronic HIV-1 infection. AIDS. 2003;17(14):2015-23.
23. UNAIDS. Gap report. Geneva: UNAIDS; 2014.
24. Al-Mahtab M, Akbar SM, Aguilar JC, Uddin MH, Khan MS, Rahman S. Therapeutic potential of a combined hepatitis B virus surface and core antigen vaccine in patients with chronic hepatitis B. Hepatol Int. 2013;7(4):981-9.
25. Aguilar JC, Lobaina Y, Muzio V, Garcia D, Penton E, Iglesias E, et al. Development of a nasal vaccine for chronic hepatitis B infection that uses the ability of hepatitis B core antigen to stimulate a strong Th1 response against hepatitis B surface antigen. Immunol Cell Biol. 2004;82(5):539-46.
26. Thio CL, Smeaton L, Saulynas M, Hwang H, Saravanan S, Kulkarni S, et al. Characterization of HIV-HBV coinfection in a multinational HIV-infected cohort. AIDS. 2013;27(2):191-201.
Received in October, 2015.
Accepted in December, 2015.
Enrique Iglesias. Departamento de Vacunas, Dirección de Investigaciones Biomédicas, Centro de Ingeniería Genética y Biotecnología. Ave. 31 e/158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.


{kind=link}
{kind=link}
{kind=link}