










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.32 no.2 La Habana Apr.-June 2015
TECHNIQUE
Scale-up of the polyacrylamide gel electrophoresis-reverse staining-extrusion-passive elution technique for the straightforward recovery of milligrams of recombinant proteins
Escalado de la técnica de electroforesis en gel de poliacrilamida-tinción inversa-extrusión-elución pasiva para la recuperación directa de milígramos de proteínas recombinantes
Eugenio Hardy1, Diógenes Quintana2, Giselle Pentón2, María del Carmen Abrahantes2
1 Laboratorio de Biotecnología, Instituto de Farmacia y Alimentos, Universidad de La Habana. Calle 222 #2317 e/ 23 y 31, La Coronela, La Lisa, CP 13600, La Habana, Cuba.
2 Centro de Ingeniería Genética y Biotecnología. Ave. 31 e/ 158 y 190, Cubanacán, Playa, PO Box 6162, CP 10600, La Habana, Cuba.
ABSTRACT
Forcing excised gel fragments through a metal sieve of 32 μm average mesh size contained in the bottom of a 1 mL syringe is a useful method to enhance passive elution of proteins. This extrusion technique works by fragmenting the gels into extremely small pieces, to maximize surface area and minimize the mean distance each protein molecule must diffuse. However, the microgel crusher is not practical when tens of 0.5-1.5-mm-thick gel slices are simultaneously processed for recovering milligram amounts of target protein. The entire procedure is time-consuming, rather tedious, labor-intensive, and requires skills. These disadvantages are also manifested when even a single, thicker (e.g., 6-12 mm) gel slice is processed for protein elution. Here, we propose solving this problem by replacing the microgel crusher with a high-speed homogenizer (Ultraturrax®, Ika). The applicability and utility of the new procedure was demonstrated in the elution and recovery of milligrams of polyacrylamide gel electrophoresis-separated pertactins (Prn). The overall elution yields were 67.6 % for Prn type 1 (6.5 mg) and 88.5 % for Prn type 2 (8.5 mg total). On average, the recovery from the process was 78 ± 14.8 % protein. As expected, the purified proteins did not show any noticeable variation in migration rate or any visible degradation products, and were detected with an anti-Prn1 monoclonal antibody (PeM19). Consequently, the new protocol will be useful for the milligram-scale isolation of target proteins separated in thick polyacrylamide gels.
Keywords: gel electrophoresis, protein, elution, pertactin, purification, recombinant protein.
RESUMEN
La extrusión de fragmentos de gel, a través de una malla metálica con un tamaño promedio de ranura de 32 μm que se encuentra insertada en el fondo de una jeringuilla de 1 mL, es un método útil para aumentar la elución pasiva de proteínas. Este método de extrusión funciona mediante la fragmentación de geles en pedazos extremadamente pequeños, para maximizar el área superficial y minimizar la distancia promedio que cada molécula de proteína tiene que difundir. Sin embargo, el microextrusor de geles no es práctico cuando decenas de fragmentos de gel de 0.5-1.5 mm se deben procesar de manera simultánea, para recuperar miligramos de la proteína blanco. El proceso completo es largo, muy tedioso, laborioso y requiere de habilidades para la manipulación del instrumental. Estas desventajas también se manifiestan cuando un fragmento de gel más grueso (por ejemplo, de 6-12 mm) se debe procesar para la elución de las proteínas que contiene. En este trabajo proponemos la solución a este problema, mediante el remplazo del microextrusor de gel por un agitador de alta velocidad (Ultraturrax®, Ika). La aplicabilidad y utilidad del nuevo procedimiento se demostró en la elución y la recuperación de milígramos de pertactinas (Prn) separadas por electroforesis en gel de poliacrilamida. Los recobrados de elución totales fueron de un 67.6 % para la Prn de tipo 1 (6.5 mg) y 88.5 % para la Prn de tipo 2 (8.5 mg total). Como promedio, el recobrado del proceso fue de 78 ± 14.8 % de proteína. Como se deseaba, el nuevo procedimiento no varió ni la movilidad electroforética ni provocó la aparición de ningún producto de degradación visible en las muestras de proteínas purificadas, y estas fueron detectadas con el uso del anticuerpo monoclonal anti-Prn 1 (PeM19). En consecuencia, el nuevo protocolo es útil para el aislamiento a la escala de milígramos, de proteínas blanco separadas en geles gruesos de electroforesis.
Palabras clave: electroforesis en gel, proteína, elución, pertactina, purificación, proteína recombinante.
INTRODUCTION
Separating and isolating recombinantly expressed proteins is critical to study and to understand their physic-chemical and biological properties. One of the most widely used techniques for this purpose continues to be slab (Laemmli’s) sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) [1]. In general, the proteins of interest are localized on the gel after SDS-PAGE and need to be eluted from the gel by several methods (e.g., passive diffusion, electroelution). Proteins eluted from gels can be used successfully in a variety of downstream applications, including protein identification and microcharacterization using immunochemical (e.g., Western blotting), biochemical (e.g., mass spectrometry) and cell-based biological methods [1, 2].
To facilitate and to enhance passive elution, we have used a micro gel crusher for fracturing small gel pieces [2]. The micro gel crusher is based on a 1-mL syringe, and has a 32-μm mesh at the bottom. The syringe piston pushes the small piece of gel through the crusher’s metal mesh, which cuts the gel into fine slurry. After soaking the gel slurry in the elution buffer of choice, the slow diffusion of proteins from the polyacrylamide matrix is accelerated by maximizing the surface area for contact between the gel and the elution buffer. Indeed, practically quantitative (90-98 %) and fast (less than 30 min) elution of proteins from SDS-PAGE gels has been achieved in the range of 1 to 100 pmol of protein per band [2].
It should be mentioned that there are other electrophoretic systems (e.g., GelFree 8100 Fractionation System, 3100 OFFGEL Fractionator) by which the sample is obtained directly in solution. These systems provide a quick, simple and reproducible method for partitioning complex protein samples into discrete molecular weight- or isoelectric point-based fractions [3]. These novel methods of fractionation eliminate the need for gel staining, band cutting and extraction from the gel sample, enabling preparative-scale fractionation of proteins in-solution with high recovery (higher than 90 %). Also, these methods are particularly useful in the recovery of low-abundance proteins, which are typically more difficult to detect, for analysis. However, compared to these other commercial Off-Gel devices, the present method has important advantages. The number of fractions is not predefined; staining the gel allows the target protein be selected a priori and separated from the gel with exquisite precision, which enables the purity of the recovered protein is significantly higher. In addition, the equipment used is available in many laboratories, and is much less expensive (e.g., GelFree 8100 costs about 16 000 USD). Finally, the reversible immobilization of the protein of interest in the gel is an aspect that facilitates largely the change of buffers and the protein unfolding/refolding process.
However, the use of the microgel crusher syringe has several disadvantages when tens of 0.5-1.5-mm-thick gel slices are simultaneously processed for recovering milligram amounts of a target protein. The entire procedure is time-consuming, rather tedious, labor intensive, and requires skills. The same disadvantages are present when even a single thicker (e.g., 6-12 mm) gel slice is processed. Because of technical difficulty in this step, a simpler and user-friendly procedure for gel crushing is desired. Here, we show the feasible replacement of the micro gel crusher by a homogenization procedure, with the aid of a rotor-stator, high-speed homogenizer (Ultraturrax®, Ika).
MATERIALS AND METHODS
Preparative SDS-PAGE
Proteins were electrophoresed in a glycine SDS-PAGE system, which used a 3.5 % stacking and a 12.5 % separating gel. The gel dimensions were 20 × 18 cm, either 6-mm- or 1.2-cm-thick, and with maximum protein loads of 10 mg or 55 mg of total protein applied onto the gels, respectively. The sample was loaded onto a wide well, followed by an electrophoresis run overnight at 20-25 mA and room temperature until the dye had reached the bottom of the gel.
Protein detection
This step was carried out as previously described [2], with modifications. Briefly, after SDS-PAGE, the gel was rinsed briefly (e.g., 30-60 s) in distilled water, and then incubated in 0.2 M imidazole solution containing 0.1 % SDS for 45-60 min. Subsequently, the imidazole solution was discarded and the gel was developed by incubation in 80 mM zinc sulfate under agitation for 30-60 s. The staining process was stopped by pouring off immediately the developing solution and washing the gel several times with abundant distilled water. By naked eyes, the PAGE-separated proteins were detected as transparent, colorless bands contrasting against a white gel background. The reverse stained protein bands were visualized better by placing the gel above any dark background.
Sometimes the staining results were improved by carrying out a double reverse staining. For this purpose, the already stained gel was incubated in 100 mM ethylenediaminetetraacetic acid (EDTA) under agitation until the white background (zinc imidazolate) deposited on the gel surface was completely dissolved, then washed with distilled water three times (each of 30 min) to remove any residual EDTA, after which the imidazole-SDS-zinc procedure was applied again to the destained gel. For gel image documentation, they were scanned using a common flatbed scanner [2]. In addition, the stained gels could be stored, without fading, in water at 4 °C for a week with reproducible results so far.
Protein passive elution
Following detection, protein bands of interest were separately excised as closely as possible with a clean scalpel and incubated (30 min) under agitation in 15-mL tubes containing 100 mM EDTA in 20 mM Tris-HCl (pH 8.0) buffer, to chelate zinc ions, and then washed (3 times for 30 min each) with 15 mL of distilled water to remove the chelating solution. Next, the excised gel fragments were transferred into 50-mL tubes loaded previously with 8 M urea in 10 mM sodium phosphate buffer (pH 7.0) and subsequently fractured with a rotor-stator homogenizer (Ultraturrax®, Ika) for maximum 10 min.
After homogenization, more elution buffer containing 8 M urea was added up to 30 mL, and the tubes with the gel slurries were placed on a platform shaker for permanent agitation at room temperature for 3 h. The tubes were centrifuged for 15 min at 1200 × g to remove the gel particles in the homogenate, and the overlaying solution was collected. This elution step was carried out four times. The solutions containing the eluted proteins resulting from each of the four elution steps were pooled, repeatedly ultrafiltered and then concentrated using Millipore’s Amicon Ultra-15 centrifugal filter devices. After this, the elution of the proteins was confirmed by a further analytical glycine-SDS-PAGE followed by gel staining with Coomassie blue R-250.
Immunodetection of PAGE-isolated recombinant pertactins
For the immunodetection of pertactin proteins (recombinant Prn 1 and Prn2, natural and native control Prn [4]), 96-wells polystyrene high binding ELISA plates (Costar® Corning Incorporated, USA) were coated for 12 h at 4 °C with 0.2 μg of either protein in 100 μL coating buffer (0.05 M Na2CO3, 0.05 M NaHCO3, pH 9.6). Subsequently, plates were washed four times with PBST buffer (10 mM sodium phosphate and 145 mM NaCl, PBS, pH 7.2, containing 0.05 % v/v Tween 20), and then blocked with 3 % w/v skim milk in PBST for 1 h at 37 °C. Next, two-fold dilutions starting at 1:2000 v/v (0.5 mg/mL) of the mAb PeM19 [5] in blocking solution were added, followed by incubation at 37 °C for 2 h and then washing (4 times) with PBST buffer. Bound mAb was detected by using horseradish peroxidase-conjugated goat anti-mouse total IgG (Sigma, St. Louis, MI). Conventional o-phenylenediamine (0.5 mg/mL)-H2O2 (0.03 %) substrate solution was used for color development; the absorbance at 492 nm was measured with a plate reader for enzyme-linked immunosorbent assay.
RESULTS AND DISCUSSION
This work was aimed to replace the microgel crusher syringe with a high-speed homogenization step (Ultraturrax® homogenizer; Ika). Hence, experimental conditions had to be established (e.g., protein load, staining time) for adequate separation and detection of proteins in thick (6-12 mm) polyacrylamide gels. To accomplish this task, we used samples with increased complexity, from single protein preparations to complex mixtures (e.g., Escherichia coli protein extract).
Figure 1 (from A to E) shows decreasing amounts of recombinant streptokinase (purified from E. coli, CIGB) separated in 6-mm thick polyacrylamide gels and stained with imidazole-SDS-zinc. The reverse-stained bands contain SDS-loaded proteins that bind zinc ions and thus prevent the precipitation of zinc imidazolate locally. Similar results were observed when the 12-mm-thick polyacrylamide gel was used (Figure 1). Various proteins from different sources were electrophoresed in SDS-PAGE and stained with imidazole-SDS-zinc to determine the general applicability of this stain. As seen in Figure 1, the P-50 protein from Serratia marcescens [6], a mixture of recombinant streptokinase and recombinant interferon α-2b, and a washed bacterial (E. coli) pellet were stained successfully by imidazole-SDS-zinc. Gels (12-mm thick) containing electrophoresed phycocianin C could be also reverse stained (data not shown).
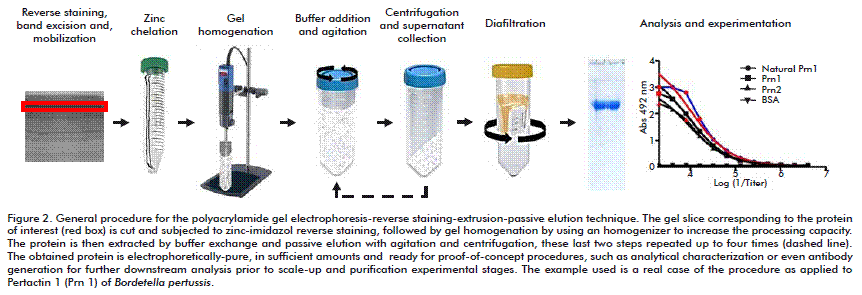
Because the gels used were significantly thicker than 1.5 mm, the reverse staining protocol had to be modified to some extent. The imidazole-SDS based equilibration step was prolonged up to 60 min, thus avoiding the subsequent development of pale reverse stained patterns. Also, after establishing the separation and detection of proteins in thick gels, we proceeded to test the proposed elution procedure, as schematically illustrated in Figure 2. Specifically, the feasibility of protein isolation on a preparative scale was demonstrated using two independent batches of complex protein mixtures containing recombinant pertactins (type 1 or 2). Pertactins are components of vaccines against Bordetella pertussis [4, 5]. The amounts obtained after each of the four elution steps were for Prn1: 1.9, 2.1, 0.33 and 2.2 mg, respectively, with an overall extraction yield of 6.5 mg (67.6 % recovery). For Prn2: 3.1, 3.6, 0.9 and 1 mg, respectively, with an overall extraction yield of 8.5 mg (88.5 % recovery).
In average, the process recovery was 78 ± 14.8 % protein. No deleterious effects such as degradation products were observed when proteins were recovered and again subjected to electrophoresis in analytical SDS-PAGE gels (e.g., Figure 2, lanes 1 and 2 for Prn 1).
Because the present study proposes a change to the already-described methodology based on gel extrusion through a mesh, it would be desirable to include a comparison of the extrusion process using an homogenizer (new proposal) to the established methodology for increased processing. The comparison should focus on the essential purpose of the method, which is to recover as much protein from the gel as possible; to this end, the recovery parameter should be used. Nevertheless, it is impossible by technical means to carry out this comparison due to the large volume of gel to be processed. Moreover, it is recommended to start the treatment at 30-50 % of performance of the homogenizer and never exceeding 70 %, to avoid dropping the tube content, or to generate any deleterious effect on the sample. The process is very mild and the result is easy to detect, since the sample readily turns into an opaque, viscous solution. The treatment may be extended for up to 10 min to guarantee the complete homogenization of gel microparticles.
Noteworthy, high elution yields have been obtained with slab-PAGE-RS-extrusion-passive elution for a variety of proteins at microgram (or less) level in a wide molecular mass range (from 21 to 97 Mr) ([2] and citations therein). In fact, when working with total radioiodinated E. coli proteins, the recovery for the 45 000-97 000 molecular mass range was 92 %. This recovery was for proteins separated in a 12.5 % PAGE gels, loaded without previous reduction, rapidly detected with imidazole-SDS-zinc and immediately eluted after the run ([2] and citations therein). In comparison to recovery at analytical scale, the values obtained at the preparative scale were slightly lower (67-88 %). Speculatively, the decrease of recovery might be related to the scaling process itself, differences in the elution buffer and additives used (SDS vs. urea), or to the solubility and other chemical-physical properties from the protein. Nevertheless, the elution at the preparative scale proved to be sufficiently high as to recover a large amount of protein, which is suitable for a variety of studies or use.
It is worth to mention that although other variants have not been evaluated, at least in theory, this method is flexible in terms of the ability to evaluate different extraction solutions for the protein of interest. Specifically, we used here 8 M urea in 10 mM sodium phosphate buffer, at pH 7.0. This elution buffer was selected due to the insoluble nature of pertactin that forms inclusion bodies when expressed in E. coli. Normally, this protein is extracted and solubilized in urea-containing extraction buffer, at pH 8.0, for unfolding/refolding and purification [4].
But it is advised to the experimenter to be careful when using elution buffers containing urea, to prevent or minimize the risk of protein carbamylation of amino groups which causes a shift on the isoelectric point of the focus protein. Proper precautions should be taken, such as: the use of a container with ice, as actually done in this work, to avoid warming of the sample above 30-37 °C during the step of homogenization of the gel and extracting the protein of interest. Also, the experimenter should i) use pure grade of urea to decrease the amount of cyanate ions present in the starting material; ii) use freshly-prepared urea solutions; iii) avoid using basic buffers as far as possible; iv) minimize the period of contact between urea and the protein; v) protein samples containing urea should be stored frozen (–20 °C) to limit cyanate accumulation; and vi) if possible, a cyanate scavenger (e.g., primary amine) should be added to the urea-containing elution solutions. If needed for subsequent use of the purified protein, it is also recommended to analyze the protein by mass spectrometry or isoelectric focusing, with the aim to detect any change generated during sample handling (e.g., carbamylation in amino groups).
Additionally, the homogenization step causes no deleterious effect on the proteins analyzed, with a reliable time window for homogenization of 10 min tops.
CONCLUSIONS
In summary, our previously described microgel crusher method [2] can be effectively scaled up with an homogenization procedure by using a conventional homogenizer (in our study an Ultraturrax® homogenizer, Ika) for enhancing passive diffusion of target proteins out of gel fragments. This procedure may find immediate, straightforward application in the milligram-scale purification of proteins that are stable even in the presence of harsh surfactants. If regaining biological (e.g., enzymatic) activity is required, the experimenter might include an in-gel renaturation step based on mild nonionic surfactant (e.g., Triton X-100), as recommended earlier [2].
The procedure is feasible and fast, been demonstrated for proteins in the range 19-72 kDa, lasting less than a month (tops two months) from cloning to obtaining an assay-ready amount of protein for proof-of-concept testings such as generating polyclonal sera for further affinity purification. And very importantly, the protein is obtained by using mild experimental procedures, without any sequence change and devoid of source contaminants, key aspects for detailed characterization studies, and at low costs at the early phases of preclinical research.
ACKNOWLEDGEMENTS
We thank Dr. Guy A M Berbers from the Laboratory for Infectious Diseases and Screening, National Institute for Public Health and the Environment, Bilthoven, The Netherlands, for kindly donating the monoclonal antibody PeM19 for pertactin proteins detection.
REFERENCES
1. Seelert H, Krause F. Preparative isolation of protein complexes and other bioparticles by elution from polyacrylamide gels. Electrophoresis. 2008;29(12):2617-36.
2. Hardy E, Castellanos-Serra LR. “Reverse-staining” of biomolecules in electrophoresis gels: analytical and micropreparative applications. Anal Biochem. 2004;328(1):1-13.
3. Witkowski C, Harkins J. Using the GELFREE 8100 Fractionation System for Molecular Weight-Based Fractionation with Liquid Phase Recovery. JoVE. 2009:34.
4. Quintana-Vázquez D, Coizeau E, Alvarez A, Delgado M, Cárdenas T, Ramos Y, et al. Recombinant hybrid proteins from pertactin type 1 and 2 of Bordetella pertussis are more immunogenic in mice than the original molecules. Biotecnol Apl. 2014;31(1):33-42. 5. Hijnen M, Mooi FR, van Gageldonk PG, Hoogerhout P, King AJ, Berbers GA. Epitope structure of the Bordetella pertussis protein P.69 pertactin, a major vaccine component and protective antigen. Infect Immun. 2004;72(7):3716-23.
6. Abrahantes-Perez MC, Reyes-Gonzalez J, Veliz Rios G, Bequet-Romero M, Gomez Riera R, Anais Gasmury C, et al. Cytotoxic proteins combined with prodigiosin obtained from Serratia marcescens have both broad and selective cytotoxic activity on tumor cells. J Chemother. 2006;18(2):172-81.
Received in February, 2015.
Accepted in September, 2015.
Eugenio Hardy. Laboratorio de Biotecnología, Instituto de Farmacia y Alimentos, Universidad de La Habana. Calle 222 #2317 e/ 23 y 31, La Coronela, La Lisa, CP 13600, La Habana, Cuba. E-mail: ehardy@ifal.uh.cu.


{kind=link}
{kind=link}