










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.32 no.4 La Habana Oct.-Dec. 2015
RESEARCH
Pre-formulation study of a pentavalent DTP-HB-Hib vaccine obtained in Ecuador
Estudio de preformulación de una vacuna pentavalente de DPT-HB-Hib producida en Ecuador
Néstor Expósito1, Eduardo Martínez1, Gladys Alvarez2, Virginia Riera2, Hugo Proaño2, Sara García2, Sara Peralta2, Diana Cevallos2, Patricia Villamar2, Jenny Navas2, Verónica Gencón2, Yayrí Prieto Correa1
1 Dirección de Desarrollo Tecnológico, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.
2 Producción de Biológicos, Empresa Pública de Fármacos, ENFARMA EP. Av. Julián Coronel 905 entre Esmeraldas y José Mascote, Guayaquil, Ecuador.
ABSTRACT
WHO has proposed the development of combined vaccines because of their advantages; in Latin America certain projects were started to this end, although the development of these vaccines is complex due to their technological challenges because of the possible negative interactions between the antigens. The National Institute of Hygiene of Ecuador asked for the collaboration of the CIGB to develop a pentavalent vaccine (DPT-HB-Hib) in Ecuador with diphtherial and tetanus anatoxin, whole cells of Bordetella pertussis (produced in Ecuador), recombinant hepatitis B virus surface antigen and the synthetic polysaccharide (polirribosyl ribitol phosphate, PRP) of Haemophilus influenzae conjugated to tetanus anatoxin (PRP-T) produced at the CIGB. A pre-formulation study was carried out; the adjuvant chosen was aluminum phosphate and the optimum absorption time was of 2h30min for the diphtherial and tetanus anatoxins and the polyribosylribitol phosphate conjugated to the tetanus anatoxin; 3h30min for the surface antigen of the recombinant hepatitis B virus and 18 h for B. pertussis. Two lots were formulated at a pilot scale with the defined technology and the potencies and immunogenicity of the antigens were assessed, as well as their identity and the percentage of adsorption, with satisfactory results. The technology for the formulation of the pentavalent vaccine produced in Ecuador was defined according to these results, which must later undergo stability studies. This is one of the first studies in Latin America related to the development of a pentavalent vaccine containing these antigens.
Keywords: pentavalent vaccine, adsorption kinetics, vaccine potency, vaccine immunogenicity, preformulation.
RESUMEN
La OMS ha propuesto el desarrollo de vacunas combinadas por las ventajas que estas ofrecen y en América Latina se han iniciado algunos proyectos. Sin embargo, su desarrollo es complejo por los retos tecnológicos que implica y que en ocasiones se evidencia en las interacciones negativas entre los antígenos. En este trabajo se desarrolló la preformulación de una vacuna pentavalente (DPT-HB-Hib) con los antígenos que incluye las anatoxinas diftérica y tetánica, células enteras de Bordetella pertussis producidas en el INH, el antígeno de superficie del virus de hepatitis B recombinante y el polisacárido sintético polirribosil ribitol fosfato de Haemophilus influenzae conjugado a la anatoxina tetánica (PRP-T) producidos en el CIGB. Se realizó un estudio de preformulación en el que se definió como adyuvante al fosfato de aluminio, los tiempos óptimos de adsorción de 2h30min para las anatoxinas y PRP-T, 3h30min para el antígeno de superficie del virus de la hepatitis B recombinante y 18 h para B. pertussis. Con la tecnología definida, se formularon dos lotes a escala piloto, a los que se evaluó la potencia y la inmunogenicidad de los antígenos, así como sus identidades antigénicas y el porcentaje de adsorción, con resultados satisfactorios. Esto permitió definir una tecnología para la formulación de la vacuna pentavalente producida en Ecuador, la cual deberá ser evaluada en estudios de estabilidad. Este es uno de los primeros estudios en América Latina para el desarrollo de una vacuna pentavalente que contiene estos antígenos.
Palabras clave: vacuna pentavalente, cinética de adsorción, potencia vacunal, inmunogenicidad vacunal, preformulación.
INTRODUCTION
The World Health Organization (WHO) and the United Nations International Children’s Fund (UNICEF), through the program World Immunization Vision and Strategy (WIVS), have established three main aims: immunize the largest number of persons against more diseases; introduce several vaccines and recently available technologies in the world; and carry out several decisive health interventions through immunization [1].
WHO currently recommends immunization routines in children against a series of infectious diseases, including diphtheria, tetanus, whooping cough, hepatitis B, poliomyelitis and invasive infections produced by Haemophilus influenzae type b [2].
The use of combined vaccines arises as a hopeful alternative. This option is conceptually practical and efficient, since it allows for the joint combination of several vaccines that are administered at the same time and at the same anatomic site, thus reducing the number of injections and simplifying the vaccination schedule [3].
The simplification of the schedule would improve its compliance, both by the parents and health professionals; there are other supplementary advantages, such as the decrease in the number of doctor’s appointments, improving transportation, storage and the decrease in the number of syringes and needles for vaccination [4]. Hence, WHO recommends the application of this type of vaccine due to its advantages [2, 5].
The combined vaccines and particularly the pentavalent DTP-HB-Hib vaccines used in different countries, made it possible for vaccines such as hepatitis B (HB) and H. influenzae type b (Hib) to be introduced in the national immunization programs without increasing the number of injections; having as its base the triple DPT.
Most of the technologies and the infrastructure needed to make these vaccines are located in industrialized countries, although there are Third World countries that can develop and produce this type of vaccine. Of course, this is a great challenge, since a combined vaccine is not the result of the simple combination of two or more antigens. It is a new vaccine and it, therefore, has to undergo regulatory inspections and its approval for marketing [6]. Additionally, technical problems may come up on combining the different antigens which may include chemical incompatibility or immunological interference. These are difficult challenges since between the antigens there may be interference resulting in the decrease of the potencies of one or all of its components and a decrease in stability. The adjuvant used, for example, can at least improve the response to one of the relevant antigens, without significantly damaging the immune responses to any other antigen of the vaccine [7-9].
Most of the interference observed in these vaccines are physical (maximum optimum potency temperature), chemical between the different components (such as the adjuvants preservatives, inactivating agents or stabilizers)[10], an incompatibility between the antigens of the DTP vaccine and those of HB and Hib, the presence or absence of thimerosal [11], the immunological and biological interference between dead bacteria or attenuated viruses and other antigens [12].
There are several pentavalent vaccine manufacturers in the world that include antigens of the diphtherial anatoxin (D), tetanus anatoxin (T), whole cells of B. pertussis (P), the surface antigen of the recombinant hepatitis B virus (HBsAg) and the polyribosylribitol phosphate conjugated to the tetanus anatoxin (PRP-T); nonetheless, the supply of these vaccines does not cover world demand, which has increased and reached 177.3 million dosages of the pentavalent vaccine in the year 2013 according to the reports of UNICEF [13].
Another problem with these vaccines is their shortage at certain times and regions, which is due to many causes, thus provoking delays or modifications in the vaccination programs, producing a deficient coverage and leading to the re-emergence of diseases [14].
This problem may, however, be minimized when a country decides to take over the challenge of the development and production of this vaccine so as to ensure its supply and a greater population coverage; this is the case of Ecuador, which already has the D, P and T antigens, through which since 1966 the National Institute of Hygiene and Tropical Medicine Leopoldo Izquieta Pérez (INHMTLIP) produces a triple DPT vaccine [15]. This DPT vaccine has been used for several decades in the National Immunization Program of Ecuador, with good results in the control of diphtheria, tetanus and whooping cough. Therefore, this triple vaccine should be the best candidate to further generate a pentavalent vaccine.
Taking advantage of the experience acquired by Center for Genetic Engineering and Biotechnology (CIGB) of Cuba since 1994 for the development and registration of combined vaccines, this work was aimed to jointly generate a pentavalent vaccine specific for the DPT antigens circulating in Ecuador, by further incorporating the HB and PRP-T antigens. The CIGB has developed and registered combined vaccines, among which are the bivalent HB-Hib, tetravalent DPT-HB, DPT-Hib, pentavalent DPT-HB+Hib and completely liquid pentavalent DPT-HB-Hib vaccines. Therefore, a joint pentavalent vaccine development project with Ecuadorian technology was implemented, to preformulate and characterize a pentavalent DTP-HB-Hib vaccine obtained in Ecuador, resulting from the implementation of a new Ecuadorean productive matrix, specifically in the production of biological agents.
MATERIALS AND METHODS
Adjuvants
To perform the different experiments we used the aluminum phosphate and aluminum hydroxide adjuvants (Adju-Phos, and Alhydrogel, respectively from the Danish company Brenntag Biosector Ltd.).
Concentration of the active pharmaceutical ingredients
In all experiments, the T antigen was used at a concentration of 20 Lf/mL, D at 50 Lf/mL, HBsAg at 20 μg/mL, PRP-T at 20 μg/mL and P at 32 opacity units (O.U.)/mL (1 opacity unit equals 109 cells), the dose used to immunize the infant population in Ecuador corresponding to 0.5 mL.
Calculation of the adsorption percentages
The adsorption percentage of each antigen in the formulations was calculated by using the equation: Adsorption (%) = (A – B × 100)/A; with A representing the amount of antigen added and B the amount of antigen identified in the supernatant of the vaccine by the specific method used for each antigen determination.
The active pharmaceutical ingredients (APIs) were obtained from two sources: the D, T and P antigens from the INHMTLIP of the Ecuadorian Republic, and the HBsAg and the PRP-T from the CIGB (Havana, Cuba). All preparations met the quality specifications.
Definition of the adjuvant in the formulation
Each D, P and T antigen were used to formulate two monovalent vaccine lots of 25 mL each; one lot was formulated in aluminum hydroxide, and the other lot in aluminum phosphate, so that each antigen was separately in contact with these two adjuvants.
The flasks having 3 % aluminum hydroxide gel and 2 % aluminum phosphate were dissolved to 1 % with 0.85 % saline solution, pH 6.6; they were sterilized at 121 °C for 15 min.
The inclusion of the antigens in each flask was carried out by continuous dripping using a Gilson P-5000 pipette. Each formulation remained under slow shaking for 1 h and the final volume was completed with 8 mM phosphate buffer saline (PBS) while continuously shaking for 15 min, and later a visual observation was made. The test was run in duplicates.
In the case of the HBsAg and the PRP-T antigens, this experiment was not carried out since the result was well documented and standardized from previous assays.
Selection of the buffer solution
The PBS was prepared at a concentration of 8 mM, (NaCl, dihydrogen dihydrated sodium phosphate, NaH2PO4 × 2 H2O and hydrogen disodium phosphate, Na2HPO4) which was added to adjust the final volume of the vaccine, reaching a concentration of between 4 and 5 mM. This procedure is used to formulate other Cuban combined vaccines such as Trivac HB® and Heberpenta-L® with very good results in the control of pH and the isotonicity of the vaccine.
These two parameters were very well controlled since this is a parenteral product, to avoid any undesirable adverse events such as pain, reddening, and induration at the application site.
Adsorption kinetics of the antigens on aluminum phosphate
Two lots of each API of D, P, T, HBsAg and PRP-T were used in this assay, to define the time needed to achieve the highest adsorption of each antigen on the aluminum phosphate gel. Five formulation variants of 25 mL each were prepared for each lot of API, except for P where we used 6 variants. This was carried out with 2 % aluminum phosphate gel that was diluted to 1 % with 0.85 % saline solution at pH 6.6; it was sterilized at 121 °C for 15 min. The APIs were added to reach the final concentration described above, while dripping with the Gilson P-5000 pipette. The adsorption process was made at with slow shaking at 200 rpm; the volume was later completed up to 25 mL with 8 mM PBS, and a 1 mL sample was taken at each sampling period.
For the APIs of D, T, HBsAg and PRP-T, the sampling periods were of 90, 150, 210, 270 and 330 min; while for the case of P these were at 3, 6, 9, 12, 18 and 24 h.
Controls
Placebos made with the same components as the above lots but without including APIs were used as negative controls, where APIs’ volumes were substituted by 0.85 % saline solution.
The positive controls were prepared with the dilutions of each API with the 0.85 saline solution until reaching the final concentration of the vaccine.
Assay for the determination of the antigen concentration in the supernatant of the vaccine
The samples were centrifuged (Hitachi SCT-15B, Tokyo, Japan) at 10 000 rpm for 5 min and 600 μL of the supernatant were then taken.
The determination of the optical density (O.D.) of each sample in the supernatant was made using the absorbance test at 280 nm in a Genesys 10UV spectrophotometer (Thermo Electron Corporation; Germany).
In the case of the measurements of PRP-T, the pentose specific Orcinol method was used to quantify this antigen, by dehydration of the D-ribose in a strongly acid medium and the later formation of the derivative, which was stained through the oxidation of this product with Orcinol in the presence of iron salts. Finally, the samples were read to determine the absorbance at a wavelength of 670 nm in the Genesys 10UV spectrophotometer (Thermo Electron Corporation; Germany).
To determine the cellular concentration of B. pertussis we used an opacity measuring method, comparing the opacity of the sample against an international reference standard recommended by WHO. Five different concentrations were prepared, these were 30, 25, 20, 15, 10 and 5 O.U./mL with 0.85 % NaCl; as a negative control we used 0.85 % NaCl.
After the established sampling periods, 2 mL of the vaccine were taken; the samples were left standing for 2 h; afterwards, and 1 mL was collected of each supernatant and compared with the standards to define the concentrations in each sample [16].
Order of inclusion of the antigens in the formulation
The antigens were added in ascending order according to molecular weight to decrease the effect of the steric impediment among them during their adsorption on the adjuvant gel.
The total volume of the gel was divided into two fractions; to one of them the following were added in this order: diphtherial anatoxin with a molecular weight of 62 kDa, and tetanus anatoxin with a molecular weight of 150 kDa. After the adsorption processes of the anatoxins, the whole cells of B. pertussis were added, with a size ranging 0.5-2.0 μm [17].
The HBsAg and the PRP-T were adsorbed in the other fraction of the total volume of the aluminum phosphate gel.
Both fractions were mixed and the buffer solution of 8 mM PBS was added until completing the final volume of the formulation, ensuring a final concentration of 4 to 5 mM PBS in the vaccine. At the end, the thimerosal preservative was added.
Formulation of two lots of the pentavalent vaccine at the scale of 1.5 L
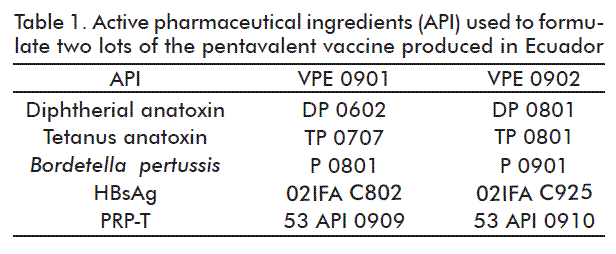
From the results obtained, we established a formulation technology for the pentavalent vaccine in Ecuador, which was assessed using physicochemical and biological trials that would show whether there was any interaction between al the antigens in the same formulation. Two lots of the pentavalent vaccine of DPT-HB-Hib were formulated at a scale of 1.5 L, labeled VPE 0901 and VPE 0902 (Table 1).
For the formulation of the two lots of the pentavalent vaccine, the 2 % aluminum phosphate gel was diluted to 1 % with 0.85 % saline solution, pH 6.6. The total volume of the diluted gel was divided into two flasks (flask 1 and flask 2); they were both sterilized at 121 °C for 15 min. In flask 1, the sterile purified diphtherial anatoxin was added at room temperature, mixing it in a magnetic shaker (IKA, RCT Basic, Germany), at 200 rpm with slow shaking for 2 h and 30 min. After this time, we added the tetanus anatoxin while still shaking for another 2 h and 30 min. Subsequently, the whole cells of B. pertussis were added and slow shaking was maintained for 18 h. In flask number 2, the PRP-T was added; the adsorption process occurred under magnetic shaking (IKA, RCT Basic, Germany), at 200 rpm the shaking was slow for 6 h; later the HBsAg was added and the adsorption process occurred during 18 h. Finally, both flasks were mixed, the 8 mM PBS was added and later thimerosal was included.
Physicochemical assays
Organoleptic characteristics
All the vials of each lot of the final product were visually inspected to verify the behavior of the sedimentation, as well as the color of the suspension.
pH determination
This method was carried out according to the requirements of the USP [18], using the equipment from Mettler Toledo AG, Switzerland.
Identification of the diphtherial and tetanus anatoxins
For the identification of the anatoxins we used the identification method of Ramon [19], consisting of mixing, under permanent observation and constant temperature, variable amounts of the anatoxin with constant amounts of the anti-toxin. The mixture that first flocculated indicated the approximate amount of anatoxin found in the sample [19, 20].
Identification of the whole cells of B. pertussis
A quantitative identification method was used, starting with the specific sera against the 3 agglutinogens, PTX, filamentous hemagglutinin and pertactin, which should be present in the B. pertussis that was obtained in the laboratory, after the immunization of rabbits with the purified antigens. The samples of the B. pertussis cells came from fractions of the two lots of the pentavalent vaccine of which we took 50 μL and mixed this with the specific sera that were previously diluted to ¼ in U-bottom Maxisorp plates (Nunc, USA). The plates were incubated for 24 h at 37 °C in a humid chamber. The wells were considered to have positive results when there was an agglutination reaction compared to a negative control where the serum was not added.
Identification of the HBsAg in the vaccine
In order to desorb the HBsAg from the adjuvant, 5 mL of the tested vaccine were taken, 400 mM PBS added and vortexed (IKA MS1, Germany) for 1 min. Later, 1 mL of the sample was centrifuged (Sigma 204, Germany) at 3500 rpm for 5 min, and 200 μL of the supernatant were collected and carried out an ELISA by covering plates with 100 μL of goat polyclonal antibody against the HBsAg (006). The plates were incubated for 15 min at 50 °C; they were then washed with 0.05 % PBS-Tween 20, three times. From the standard solution and from the vaccine, 200 μL each were taken and diluted twice with PBS; the samples were applied to the plate and incubated for 4 h at 37 °C in a humid chamber. They were washed as mentioned above and 100 μL of the anti-goat peroxidase conjugate (1/100) were added to the plate. They were incubated for 1 h at 50 °C, washed with 100 μL of the citrate-phosphate buffer solution containing the ortho-phenylendiamine substrate at 1 mg/mL and 30 % H2O2.
The reaction was stopped by adding 50 μL of the stop solution (2 M H2SO4), and the plate was read at 492 nm in a Titertek Multiskan Plus plate reader (Labsystems, Finland). The results were considered as positive if the HBsAg was identified in the vaccine tested.
Identification of the PRP-T in the vaccine
The qualitative method for immune-identification by latex was used through a commercial set of reagents Pastorex ® Meningitis (61716). In this determination, a drop of the well homogenized vaccine was taken with a Pasteur pipette, further mixed with a drop of the latex sensitized with rabbit monoclonal antibodies specifically against the capsular polysaccharide of the H. influenzae type b bacterium. The test was considered as positive if the sample agglutinated the positive control equally.
Adsorption of the antigens in the vaccine
The amounts of the diphtherial and tetanus anatoxins were quantified by the Ramon method [19]; the valuation of the B. pertussis cellular concentration was determined using the opacity meter method [16].
HBsAg was quantified by the ELISA method described above; for the determination of the PRP-T we were not able to use the Orcinol method since the B. pertussis is a source of ribose units, reporting higher values in the final result of the measurement. Therefore, the value was assumed to be that determined in adsorption assays for the PRP-T antigen alone.
Western blot
Since the antigen mix found in pentavalent vaccines complicates the determination of HBsAg by using electrophoresis under normal conditions, the two lots of vaccine were tested with a variation of the technique, also assessing if there were any interactions between this antigen and the rest of the components of the formulation. Electrophoresis was run at a gradient of 5-15 % and the current was of 15 mA until it was linearized at 30 mA, followed by a fast staining with Coomassie G-250 . Subsequently, Western Blot was carried out as described [21]. The electrophoresis was transferred to the nitrocellulose membrane with a current of 1.00 mA for 1 h using a humid transfer. It was blocked for 2 h at 37 °C with 1× PBS, 0.05 % Tween-20 and 5 % milk (w/v). After blocking, the membrane was washed twice with 1× PBS, 0.05 % Tween-20 and it was incubated with the MAb Hep.1/peroxidase conjugate in 1× PBS, 0.05 %, Tween-20 and 1 % milk at 37 °C for 2 h. Afterwards, the membrane was washed three times with 1× PBS, 0.05 % Tween-20 and developed with diamine benzidine (DAB) and hydrogen peroxide (H2O2).
Biological assays
Procedure for the determination of the potency of the diphtherial anatoxin according to the indirect method 1 (FDA)
This procedure is based on the neutralization capacity of the diphtherial anti-toxin found in the serum of animals that had been immunized with the pentavalent vaccine when facing a reference toxin [22].
The reference diphtherial anti-toxin was diluted up to 0.5 IU/mL. A 1-mL sample of the animal sera mixture was taken and poured into a tube containing 9 mL of the 0.85 % saline solution (dilution/10); the previous operation was repeated in a tube containing 19 mL of the 0.85 % saline solution (dilution 1/20). Five tubes were kept in the dark at 37 ºC for 4 h, containing 1 mL of the diphtherial toxin each, at the dilutions of the serum and the Glenny buffer of 4 mL/1 mL, 2mL/2 mL, 1 mL/3 mL, 0.5 mL/3.5 mL and 0.25 mL/ 3.75 mL.
In two guinea pigs of 500-700 g, their upper parts were divided into squares up to 15 squares. The animals were inoculated with 0.1 mL of each preparation through the intradermal route. They were observed at 24 and 48 h. Using a slide gauge the erythemas of the animals were measured and the diphtherial anti-toxin content (IU/mL) in the sera mixture was determined.
The lot passed the test if there were at least 2 IU of the diphtherial anti-toxin per milliliter of the serum [22].
Procedure used to determine the potency of the tetanus anatoxin according to the Indirect method 1 (FDA)
Two milliliters of the pentavalent vaccine were taken and 11.3 mL of the sterile 0.85 % saline solution were added. Ten guinea pigs of the Hartley line of 250 to 350 g of weight were immunized with 1.0 mL of the above dilution. Four weeks later, they were given an equal dose and 15 days afterwards the animals were bled. The test tubes were incubated at 37 °C for 2 h; thereafter, the clots were separated from the walls of the tubes and were refrigerated at 5 ± 3 °C to retract the clot.
From each animal, 1 mL of the serum was collected and they were pooled in a test tube. For the titration of the tetanus anti-toxin, 60 mice of the same sex were used, weighing 16 to 18 g; 30 mice were used for the reference system and 30 for the problem system (pentavalent vaccine). The problem serum was diluted 1/30 with 0.85 % saline solution. For the preparation of the tetanus toxin, 10.868 mg of the toxin were weighed and diluted in 22 mL of 0.85 % saline solution.
The anti-toxin standard was prepared having 4 IU/ mg of anti-toxin, by weighing 1.5 mg of the anti-toxin and diluting it in 6 mL of saline solution, obtaining a standard system at 1 IU/mL.
The solutions were mixed and placed in the dark for 1 h at between 20 and 25 °C. Afterwards, mice were inoculated with 0.5 mL of the mixture per animal by subcutaneous route, and observations were made at 24, 48, 72, 96 and 120 h.
The titer of the vaccine was obtained by using the statistical method of Sperman and Karber. The dilution at which all animals survived was taken into account, which corresponded with the total mortality and the intermediate value between them. With the above mentioned data, the ED50 was calculated for the standard and the problem sera.
The test was considered to be satisfactory in the analysis of a vaccine lot if the titer obtained was at least 2 IU of the tetanus anti-toxin per milliliter of serum [23].
Procedure for the determination of the potency of B. pertussis using the method recommended by WHO
The B. pertussis potency of the vaccine was determined by the comparison to a working reference vaccine calibrated against the international standard for the P vaccine approved by the Quality Control Division of BioCen, Cuba.
Four dilutions of the reference vaccine were made and from each lot of the vaccine to be tested. The serial dilutions were made with a dilution factor that was no larger than five, using for this the sterile 0.85 % sodium chloride saline solution.
Albino OF-1 mice of 10-18 g of weight were injected intraperitoneally with 0.5 mL of the dilution corresponding to each mouse in each immunization group. Later, mice immunized with the reference vaccine and the trial vaccine were injected with the challenge dose by intracerebral route at a time interval of 14-17 days after immunization. The strain used for the challenge was B. pertussis 18 323.
To obtain an estimate of the LD50, we carried out dilutions of the challenge dose (1/50, 1/250, 1/1250) which were inoculated by the intracerebral route in control mice groups.
The appropriate dilutions of the challenge dose were grown in a Bordet-Gengou agar base to determine the number of colony forming units (c.f.u.). The animals were observed for 14 days; the mice that died within the first 72 h after the inoculation were excluded from the assay. The mice dying after 72 h of the inoculation were recorded to determine the ED50 of the vaccines. The ED50 was determined for each preparation using the Probit statistical method, which evaluates the linearity of the dose-response and the parallelism of the behavior of the tested vaccine with the reference vaccine.
The value of the ED50 of each vaccine is the intermediate value between the highest and the lowest immunizing dose and the regressions that did not show significant deviations from linearity and parallelism (p ≤ 0.05). The challenge dose contained between 100-1000 LD50 and no more than 300 c.f.u.
The ED50 of the vaccine under trial and the standard vaccine were calculated by a method that offers an estimate of the 95 % confidence interval limits. The potency was estimated in terms of IU in the recommended volume for a single human dose (SHD).
The vaccine in the trial met the requirements for the potency if the result of the trial was statistically valid, showing that the estimated potency of the vaccine was not less than 4.0 IU in the volume recommended for an SHD, and if the lower limit (p = 0.95) of the estimated potency was not less than 2.0 IU [24].
Determination of the relative in vivo potency of the HBsAg
The potency test of the HBsAg was carried out according to the technical requirements of WHO [25].
One milliliter of the trial vaccine was taken, which contained 20 μg/mL of HBsAg; it was diluted 1/16, 1/64, 1/256, 1/512 and 1/1024 with aluminum phosphate gel at a concentration of 0.5 mg of Al+3/mL.
Ten female mice of 5 to 6 weeks of age and of the Balb/c, haplotype H-2d,q were immunized per group, using the intra-peritoneal route. Two lots of the pentavalent vaccine, one lot of the placebo and the reference lot of the anti-hepatitis B vaccine, 07-0902, were studied.
Twenty-eight days later, the mice were bled by the retro-orbital route and the HBsAg antibody response was evaluated using an ELISA system; for this, it was coated with HBsAg (solid phase), the sample was incubated in the wells of the plate and then the HBsAg conjugated with hot radish peroxidase was added. Ortho-phenyldiamine was added as the chromogenic substrate to develop the reaction. The ELISA plate was read at 492 nm.
The lot passed the test when the value of the relative potency equaled or was larger than 0.5 compared to the potency of the reference vaccine.
Determination of the immunogenicity of the PRP-T component
Groups of five F1 rabbits were used, which were immunized by the subcutaneous route with the dose of 10 μg of the pentavalent vaccine. At the same time, the rabbits were immunized with a control vaccine against Hib (Vaxem Hib, Chiron S.p.a, lot 3204), and an aluminum phosphate placebo (0.5 mg/mL) was used as the negative control.
The rabbits were immunized at 0 and 14 days and they were bled at 21 days after the first dose. Blood was collected individually.
The response to antibodies was assessed by a specific ELISA system for PRP-T that was non-competitive and indirect. The international recommendation was used in the coating, i.e., the capsular polysaccharide of the bacterium covalently conjugated to human serum albumin (HbO-HA), distributed by NIBSC, England. The addition of the sample was carried out to form the HbO-HA+Ac anti-PRP-T complex. This complex was bound to the mouse anti-IgG conjugate marked with peroxidase. Orthophenylendiamine was used as the chromogen; the substrate of the reaction was H2O2. In the positive antibody samples there was a yellow-orange coloring.
For each one of the samples of the trial we calculated the percentage of seroconverted animals for each dose; the lot passed the test when seroconversion was found at least in 50 % of the animals immunized per study group, and the average of the titers of each group of animals immunized with the pentavalent vaccine was equal or higher than 800 IU/mL.
Statistical analysis
For the statistical analysis of the different values in the adsorption kinetics study, the ANOVA F-test was applied to determine if there were any significant differences between the means of the samples with a 95.0 % confidence interval. If this was true, the Multiple Range Test was applied to define the means that were significantly different from the others, using the Fisher’s least significant difference (LSD) method. There was a risk of 5.0 % of saying that each pair of means is significantly different, when the true difference equals 0. For this analysis, we used the STATGRAPHICS Centurion XV.II statistical program.
RESULTS AND DISCUSSION
Definition of the adjuvant to be used in the pentavalent vaccine formulation
The objective of the assay was to evaluate the organoleptic characteristics of these vaccine preparations. According to the results obtained in this experiment, we defined the aluminum phosphate gel as the adjuvant, because the whole cells of B. pertussis produced in Ecuador is inactivated with thimerosal, this preservative therefore incompatible with the aluminum hydroxide gel due to the formation of lumps, as observed in figure 1.
This phenomenon may be caused by B. pertussis cell wall adhesins, such as filamentous hemagglutinin, pertactin and pertussis toxin, which in the presence of heat or thimerosal can favor the cell agglutination process, so that when combined with the aluminum hydroxide, the lumps are produced. For instance, in the case of the Cuban pentavalent vaccine, the aluminum hydroxide is used because the P is inactivated with formaldehyde that does not favor the formation of lumps with the gel. Regarding the D and T antigens, in the presence of aluminum hydroxide or aluminum phosphate, do not show undesirable interactions as seen in figure 1.
No experiment was carried out to define the adjuvant for HBsAg and the PRP-T, because previous experiments have shown that there is no interaction in neither of them with both adjuvants.
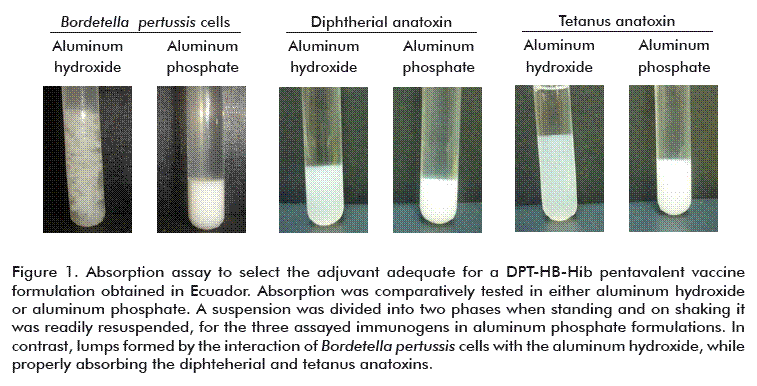
The aluminum phosphate gel has a zeta potential (ZP) of 5.0, i.e., above pH 5.0 its charge is predominantly negative; that is why it is designed to absorb antigens with high isoelectric points, when they have predominantly positive charges. The selection of the aluminum phosphate gel for the formulation of this pentavalent vaccine predicts the discrete adsorption values for the different antigens, according to their isoelectric points (Ip). For example, that of HBsAg is 4.5, the anatoxin D has 4.1 and T is 5.1 [26]; so that the predominant charge of these antigens is negative, similar to that of the aluminum phosphate when the pH is near the neutrality value, as observed in Figure 2. Overall, this means that a negative electrostatic effect is expressed. The contrary occurs when using the aluminum hydroxide, since near neutrality the hydroxide has a positive charge, in contrast to the HBsAg, establishing a positive electrostatic effect that favors the adsorption process.
In previous adsorption experiments using an aluminum phosphate gel, we have obtained values of about 80 % for the HBsAg and of about 40 % for the PRP-T (unpublished results).
Definition of the buffer solution for the formulation of the pentavalent vaccine
Salts are fundamental elements in the composition of vaccines; they fulfill different functions such as the regulation of isotonicity of the vaccine preparation and pH.
These two parameters should be well controlled in a parenteral product, since when uncontrolled, they can produce the rupture of muscular tissue because of the difference in the salt composition and the pH, giving place to undesirable adverse events such as pain, reddening, and induration at the application site.
On the other hand, the degree of adsorption of the antigens depends on their nature and concentration, on the presence of salts and ions such as buffers and on the pH of the resulting mixture [27]; these elements are achieved using the PBS 8.0 mM in the formulation.
Many buffer solutions can be used in the formulation of a vaccine, but our experience shows that we have had good results in other vaccines using PBS 8.0 mM (NaCl, dihydrogen dihydrated sodium phosphate, NaH2PO4 · 2H2O and disodium hydrogen phosphate, Na2HPO4) which is used to complete the final volume of the vaccine, with a final concentration of between 4.0 and 5.0 mM.
Adsorption kinetics of the antigens in the aluminum phosphate
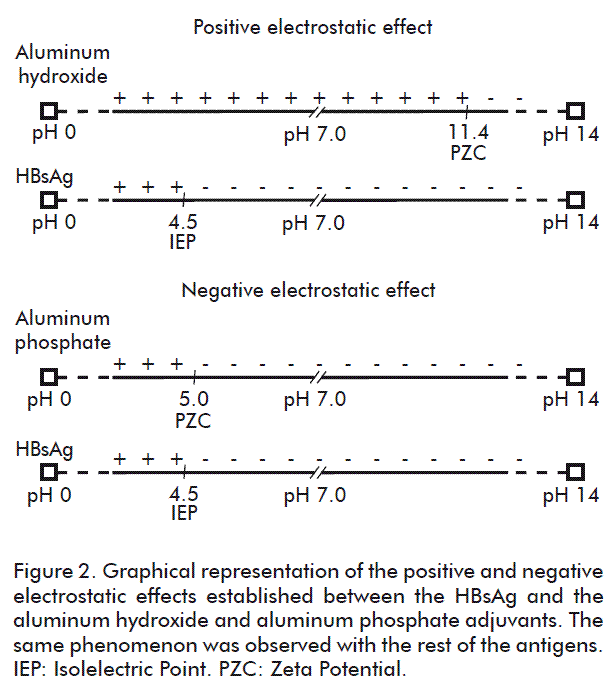
The objective of this study was to define the time needed to reach the highest adsorption of each antigen onto the aluminum phosphate gel. The results of the kinetics of adsorption of D in the aluminum phosphate are shown in Figure 3 A. The adsorption kinetics of the diphtherial anatoxin shows that 2h30min is enough to reach the maximum adsorption of this antigen, since the results show no significant differences.
As observed between the values of the quantified diphtherial anatoxin O.D. in the supernatant of the vaccine, at the time periods of 1h30min and 2h30min, there is a significant difference, but between the values reached at 2h30min and the rest of the values corresponding to longer time periods, there were no significant differences. This indicates that the time for a maximum adsorption of this antigen to the aluminum phosphate adjuvant under these conditions is of 2h30min.
The results of the adsorption kinetics of T to the aluminum phosphate gel are shown in Figure 3 B. A similar result was reached with the tetanus anatoxin, with 2h30min as enough to achieve the highest adsorption of this antigen onto the aluminum phosphate, without any significant difference between this result and those reached at longer periods.
The behavior of the dynamics of the adsorption of T is similar to that obtained with D; as of 2h30min the maximum adsorption is reached and there are no significant differences in the values as of this time in relation to the results of longer periods.
The kinetics of the adsorption of whole cell B. pertussis was a much slower process, which may be observed in Figure 3 C; here regardless of the fact that these cells present on their cell wall a chemical composition based mainly on proteins favoring the interaction with the aluminum adjuvants, [28] their size influenced the adsorption process.
After 3 h, there were approximately 30 O.U./mL of B. pertussis whole cells in the supernatant ,of the total added. At 6 h, this was of 25 O.U./mL, showing that this adsorption process is occurring, since at 9 and 12 h the quantification was of 15 and 10 O.U./ mL, respectively; is process, however, was able to conclude as of 18 h since the result of 24 h is similar, at which 5 O.U./mL were quantified.
This result was similar in both APIs of B. pertussis; because of this only one curve is shown in the graph indicating that the process concludes at 18 h of adsorption, where up to 5 O.U./mL were adsorbed, and this was sufficient time, since the result reached at 24 h is similar to that at 18 h. All results quantified in the different times showed significant differences except for 18 and 24 h. This shows that for completing the adsorption process, 18 h are required. It is important to point out that the behavior was identical in both pertussis lots and because of this the graph only shows one curve although both lots are represented.
The adsorption kinetics of the HBsAg is shown in Figure 3D. This antigen presents certain physic-chemical characteristics that give it a predominantly negative charge at a pH that is close to neutrality, similar to that of the aluminum phosphate; therefore, the adsorption process does not occur as fast as in the case of the adsorption to the aluminum hydroxide, where it is facilitated by a ligand exchange process [29].
Nonetheless, the adsorption process basically occurs by hydrophobic interactions. It should be observed how most of the adsorption occurs in 3 h and a half and the results after this time period do not show significant differences.
The results of the adsorption kinetics of the PRP-T are shown in Figure 3 E. This process indicates that the largest amount of this antigen absorbs onto the aluminum phosphate at 2h30min, representing approximately 40 % of the total PRP-T added. It is evident that a negative electrostatic effect is expressed, as occurring with the rest of the antigens with charges that are opposite to those of the gel, and, therefore, the predominating interactions are the hydrophobic interactions.
Adsorption experiments of the PRP-T onto the aluminum phosphate at pH 3 to 4 have been carried out, with percentages of adsorption above 97 %. But, when the pH was increased to reach a value that was near neutrality, more than half of the PRP-T in the vaccine de-adsorbed from the aluminum phosphate, reaching only 40 % of PRP-T adsorbed. This event was caused by the increase of the H+ ion concentration of HCl, since this ion is small and can readily bind to the negative surface charges of the gel, thereby inverting the net load of the aluminum phosphate from negative to positive. Hence, the electrostatic interaction between the PRP-T and the aluminum phosphate is facilitated. In this process of increasing the vaccine pH up to 6.85, 0.2 M NaOH was added, the OH- ions displacing by size the PRP-T previously adsorbed. Therefore, this antigen becomes de-adsorbed as shown in figure 3 F.
Other studies on the adsorption of antigens to aluminum gels have demonstrated that the process is mediated by electrostatic, hydrophobic and ligand exchange interactions, all of them contributing to the adsorption of the given antigen to the adjuvant by their respective mechanisms [30, 31]. Nevertheless, a lower or higher percentage of adsorption of an antigen depends on their physicochemical characteristics, the type of interactions achieved according to the pH of the medium and the net charge of the adjuvant under the test conditions.
As a general rule, for several antigens, the adsorption process is performed more effectively in the pH interval where the isoelectric point of the antigen and the zero zeta potential of the adjuvant have opposite charges, as shown in Figure 2. This condition is met both for the phosphate and for the aluminum hydroxide. In fact, this is interval at which the adjuvant and the antigen have opposite electric charges that facilitate the electrostatic attraction, the ligand exchange and the adsorption process.
Order in which the antigens are added
The inclusion in the formulation of the diphtherial anatoxin as the first antigen is sustained, because it is much less immunogenic and it is a substantially smaller molecule compared to the rest of the antigens of the vaccine. Epitopic localization studies have shown that the largest epitope protector found in the diphtherial anatoxin is discontinuous and small in extension; hence it is more susceptible to damage or masking during the technological processes and it is therefore less competitive when presented to the histocompatibility cells [32, 33].
On the other hand, it has been verified that the percentage of adsorption of the diphtherial and tetanus anatoxins vary when bivalent vaccines against diphtheria-tetanus are formulated. Here the adsorption of these antigens to the adjuvant gel is higher than when B. pertussis is incorporated to produce the triple vaccine, DPT. This suggests that these cells displace the anatoxins from the sites of the adsorption in the gel, which is considered as one of the potential problems associated to combination vaccines [16].
In the case of the HBsAg and the PRP-T, they were adsorbed in parallel on a fraction of the total volume of the adjuvant gel. This was aimed to minimizing the possible interactions with D, P and T the PRP-T and the HBsAg, so that on mixing both adsorptions, a large part of the antigens could be already mobilized in the matrix of the adjuvant, thus ensuring a greater protection and decreasing the interaction between them.
Physicochemical tests of the two lots of the pentavalent vaccine
Organoleptic characteristics
For both lots of the pentavalent vaccine, a grey-white suspension was obtained, which was separated into two phases after standing for a certain time, yielding a transparent liquid supernatant and a precipitate that corresponded to the adjuvant gel. After shaking the vaccine, it was readily resuspended, showing a homogeneous suspension.
pH
After concluding the formulation, both lots (VPE 0901 and VPE 0902) had pH 6.6. After 6 months of storage, the pH of the lots had similar values, of 6.6 and 6.7 respectively. This demonstrated that the 8 mM PBS solution guaranteed this parameter at least for the time evaluated, and at 5 ± 3 ºC.
Identification of the antigens in the formulation
The results of the identification of the antigens in the two lots formulated for the pentavalent vaccine are shown in Table 2, with identity values as established for anatoxins. This result supports the production of both antigens with the adequate potency, since this parameter is directly related to the capacity of the preparation to induce an immune response in animals, which in the case of D and T highly correlates with seroprotection in humans.
The whole cells of B. pertussis, HBsAg and PRP-T were identified in both lots. Regardless of using specific antibodies to identify the five antigens, the biological tests provided additional information that may confirm that there is no interference between the antigens, excipients, preservatives and adjuvants in the pentavalent vaccine, at least in the first stage.
Adsorption of the antigens in both lots of the pentavalent vaccine
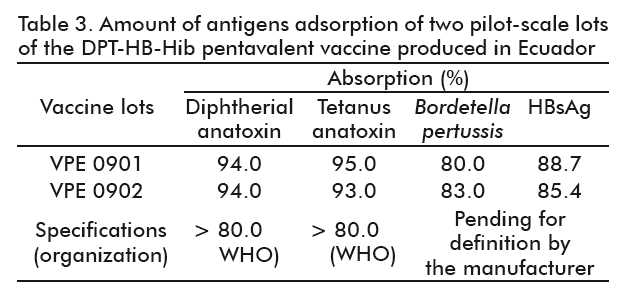
The results of the percentage of adsorption of the D, T, HBsAg and P in both lots of the pentavalent vaccine are shown in Table 3. Results for D and T in both lots were above 93 %, complying with WHO recommendations stating that vaccines containing these antigens should be adsorbed in more than 80 % [34].
This positive result directly derived from the inclusion order of the anatoxins, the defined adsorption periods according to the kinetic studies and the use of 8 mM PBS. This last remains at a final concentration from 4 to 5 mM after completing the final volume of the vaccine, thereby, ensuring the pH of the vaccine in the medium.
The adsorption of the whole cells of B. pertussis in the pentavalent vaccine was obtained at 80-83 % for the VPE 0901 and VPE0902 lots, similar to those values reached in kinetic studies of monovalent pertussis vaccines. Hence, it was demonstrated that there was no interference for this parameter on combining the five antigens. Noteworthy, there are no WHO recommendations regarding the percentage of adsorption of the whole cells of B. pertussis to aluminum adjuvants.
HBsAg was able to be adsorbed at 85 to 87 %, which is a positive result considering the conditions generating opposite charges between this antigen and the aluminum phosphate that do not favor this process. In fact, WHO recommends that the degree of adsorption for this antigen should be evaluated in each lot of the vaccine, and that specifications or ranges for approval have to be established once demonstrated the consistency of the results evaluated for the approval of the National Regulatory Authority [25].
Western Blot anti-HBsAg
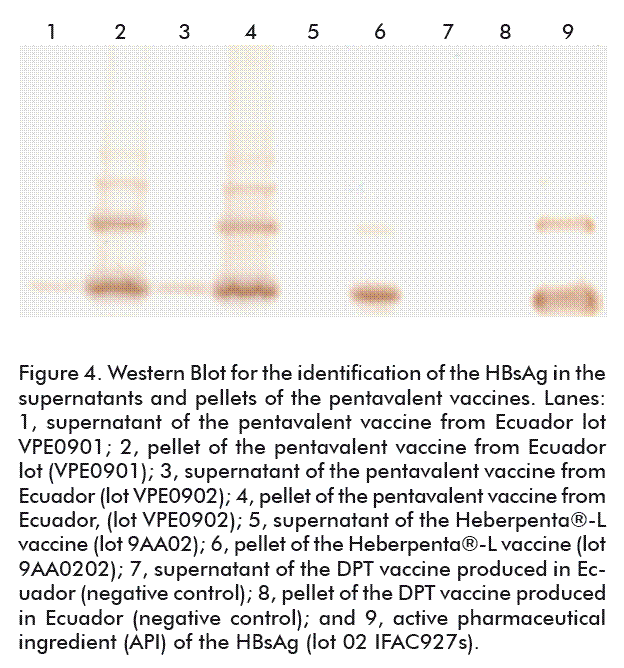
A more specific test other than electrophoresis had to be made to detect the presence of this antigen in both vaccine lots, for a clear identification of HBsAg on the pentavalent vaccines. Additionally, it was evaluated if there were any interactions between this antigen and the other formulation components.
Figure 4 shows the anti-HBsAg Western Blot made on samples of supernatants and pellets of both pentavalent vaccine lots, VPE 0901 and VPE 0901. As observed in lanes 1 and 3, corresponding to vaccine supernatants, HBsAg was effectively detected as expected, since the percentage of absorption for this antigen in the respective test rendered 11.3 to 14.6 % of the antigen unabsorbed.
As shown in lanes 2 and 4, corresponding to the pellets of this same vaccine, there were bands with greater intensities corresponding to aggregates, such as HBsAg dimers and trimers with 88.7 and 85.4 % of this antigen adsorbed onto the aluminum phosphate gel, respectively.
In the case of supernatant and pellet of the pentavalent vaccine lot 9AA0202 produced in Cuba, lanes 5 and 6, a different pattern was obtained, since the HBsAg was not identified in the supernatant but to 100 % in the pellet, because of using the aluminum hydroxide gel with which the interaction is established by a very strong ligand exchange interaction. No specific bands were present in lanes 7 and 8 (supernatant and pellet, respectively) corresponding to the DPT vaccine obtained in Ecuador, devoid of HBsAg, while detected for the positive control of an HBsAg-containing API (lane 9), with clear evidences of the monomer and dimer molecular species of that molecule.
According to these results, there is no interference between HBsAg and the other antigens in the formulation, indicated by absence of degradation.
Potency and immunogenicity of the antigens in the lots of the pentavalent vaccine
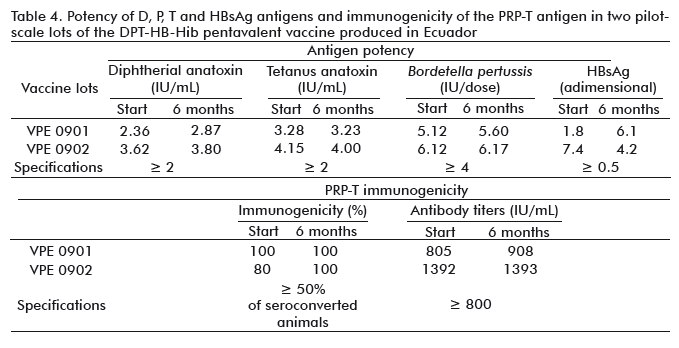
Potency and immunogenicity results for the five antigens in both vaccine lots complied with the specifications established for the study, as shown in Table 4, at both time periods evaluated.
The combination of several antigens of different nature within the same formulation is always a challenge, since there could be negative interactions that could affect the potency or immunogenicity of the antigens, considering that the HBsAg comes from a virus and that the PRP-T is synthetically obtained. The results of the potency of the HBsAg were very much higher than the specification established, i.e., the response of the lots of the pentavalent vaccine produced in Ecuador was several times higher for the HBsAg than the control lot, which is a monovalent vaccine. This could be related to the immunomodulating properties of some B. pertussis components which contribute with immunostimulatory effects on the immune response for some of the antigens in vaccine preparations [35].
PRP-T results demonstrated that at least in the time period evaluated, there was a good response since more than 80 % of the animals seroconverted and the titers obtained were higher than 800 IU/mL [36].
The variability observed is characteristic for this type of biological assay [31]. The methods to determine potency and immunogenicity are frequently highly variable, due to variations in the biological response in animals and during sample handling for experimental determinations.
In summary, the obtained potency and immunogenicity results demonstrated no immunological interferences, either chemical or between the antigens, the adjuvant or thimerosal preservative during animal testing, one of the main purposes pursued at the pre-formulation stage.
CONCLUSIONS
It was possible to define a technology for the production of a DPT-HB-Hib pentavalent vaccine in Ecuador at the preformulation stage, comprising the selection of aluminum phosphate as adjuvant, establishing the adequate order in which the antigens are added, setting the optimal absorption time period to 2h30min for the diphtherial and tetanus anatoxins and the PRP-T, and 3h30 min for HBsAg and 18 h for whole cell B. pertussis.
Satisfactory results were obtained in the physico-chemical testing for pH, the identity of antigens, with high adsorption percentage and adequate organoleptic characteristics established for two pentavalent vaccine lots formulated at pilot scale. Potency and immunogenicity of the five antigens present in the formulation were adequate as well.
REFERENCES
1. Expósito NS, Cardoso D, Martínez E, Herrera Y, Cosme K, Díaz PA, et al. Vacuna combinada cubana Trivac HB®, Biotecnol Apl. 2006;23(2):158-64.
2. World Health Organization. WHO. Recommendations for Routine immnunization – summary tables. 2012 [cited 2015 Aug 16]. Available from: http://www.who.int/immunization/policy/immunization_tables/en/
3. Schmitt HJ, Booy R, Weil-Olivier C, Van Damme P, Cohen R, Peltola H. Child vaccination policies in Europe: a report from the Summits of Independent European Vaccination Experts. Lancet Infect Dis. 2003;3(2):103-8.
4. Martín-Moreno JM. De vacunas combinadas pentavalentes a hexavalentes: esperar o no esperar, esa es la cuestión. JANO. 2005;(1589):63.
5. Skibinski DA, Baudner BC, Singh M, O’Hagan DT. Combination vaccines. J Glob Infect Dis. 2011;3(1):63-72.
6. US Food and Drug administration. Guidance for industry for the evaluation of combination vaccines for preventable diseases: production, testing and clinical studies; 1997 [cited 2015 Aug 16]. Available from: http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/UCM175909.pdf
7. Halsey NA. Safety of combination vaccines: perception versus reality. Pediatr Infect Dis J 2001;20(11 Suppl):S40-4.
8. Yeh SH, Ward JI, Partridge S, Marcy SM, Lee H, Jing J, et al. Safety and immunogenicity of a pentavalent diphtheria, tetanus, pertussis, hepatitis B and polio combination vaccine in infants. Pediatr Infect Dis J. 2001;20(10):973-80.
9. Elliman D, Bedford H. Safety and efficacy of combination vaccines. BMJ. 2003;326(7397):995-6.
10. Vose J. Regulatory considerations for developers. In: Ronald W. Ellis, ed. Combination vaccines. Totowa, NJ: Human Press; 1999. p. 213-33.
11. Insel R. Potential alterations in immunogenicity by combining or simultaneously administering vaccine components. Ann N Y Acad Sci. 1995;754:35-47.
12. Falk LA, Arciniega J, McVittie L. Manufacturing issues with combining different antigens: a regulatory perspective. Clin Infect Dis. 2001;33 Suppl 4:S351-5.
13. Unicef. Influencing markets to reduce vaccine prices. 2015 [cited 2015 Aug 16]. Available from: http://sowc2015.unicef.org/stories/influencing-markets-to-reduce-vaccine-prices/
14. Meeting of the Strategic Advisory Group of Experts on Immunization, November 2011 - conclusions and recommendations. Wkly Epidemiol Rec. 2012;87(1):1-16.
15. Aguas Ortiz JC. La Salud Pública en el Ecuador en el siglo XX: El Instituto Nacional de Higiene y Medicina Tropical “Leopoldo Izquieta Pérez” (INHMT-LIP, 1937-1980). Barcelona: Universidad Autónoma de Barcelona; 2012.
16. OMS. Manual para la producción y control de vacunas Bordetella pertussis; 1979.
17. Galazka AM. The immunological basis for immunization series. Module 2. Diphtheria. Geneva: World Health Organization; 1996.
18. United States Pharmacopoeial Convention. United States Pharmacopeia 29, 2006. National Formulary 25. Rockville: United States Pharmacopoeial Convention, Inc. 2006.
19. Ramon G. Floculation dans un élange neutre de toxin-antitoxine diphtériques. C R Soc Biol. 1922;86:6.
20. World Health Organization. Manual for the Production and Control of Vaccines: Tetanus Toxoid; 1978.
21. PPO 1.34.602.03. Procedimiento para el reconocimiento inmunológico por electrotransfer de proteínas específicas o de la cepa hospedera en productos en desarrollo. La Habana: CIGB; 2011.
22. PNO 07.153. Procedimiento establecido para determinar la potencia de la anatoxina diftérica en el BioCen. La Habana: BioCen; 2015.
23. PNO 07.161. Procedimiento establecido para determinar la potencia de la anatoxina tetánica en el BioCen. La Habana: BioCen; 2014.
24. BioCen, 2014. PNO 07.160. Procedimiento establecido para determinar la potencia de la Bordetella pertussis en el BioCen. La Habana: BioCen; 2014.
25. WHO. Recommendations to Assure the Quality, Safety and Efficacy of Recombinant Hepatitis B Vaccines. WHO Technical Report Series. 2013;(978):189-240.
26. Pillemer L, Wittler RG, Burrell JI, Grossberg DB. The Immunochemistry of toxins and toxoids. VI. The crystallization and characterization of tetanus toxin. J Exp Med. 1948;88:205-21.
27. Clapp T, Siebert P, Chen D, Jones Braun L. Vaccines with aluminum-containing adjuvants: optimizing vaccine efficacy and thermal stability. J Pharm Sci. 2011;100(2):388-401.
28. Gilchrist M. Bordetella. In: Balows A, Hausler WJ, Herrmann KL, Isenberg HD, Shadomy HJ, eds. Manual of clinical microbiology, 5th ed. Washington DC: American Society for Microbiology; 1991. p. 471-7.
29. Vogel FR, Hem SL. Immunologic adjuvants. In: Plotkin SA, Orenstein MD, eds. Vaccines. 4th ed. New York: Saunders; 2004. p. 69-79.
30. Morefield, GL, Jiang D, Romero IZ, Geahlen RL, Hogenesch H, Hem SL. Effect of phosphorylation of ovalbumin on adsorption by aluminum-containing adjuvants and elution upon exposure to interstitial fluid. Vaccine. 2005;23(12):1502-6.
31. Bleam WF, Pfeffer PE, Goldberg S, Taylor RW, Dudley RA. 31 P solid-state nuclear magnetic resonance study of phosphate adsorption at the boehmite/aqueous solution interface. Langmuir. 1991;7:1702-12.
32. OMS. Guía de la OMS. Requisitos de las prácticas adecuadas de fabricación (PAF). WHO/VSQ/97.01. Ginebra: Organización Mundial de la Salud; 1998.
33. Corbel MJ. Control testing of combined vaccines: A consideration of potential problems and approaches. Biologicals. 1994;22(4):353-60.
34. EMEA. Committee for Proprietary Medicinal Products, European. Note for Guidance on Pharmaceutical and Biological aspects of combined -vaccines. London: Agency for the Evaluation of Medicinal Products; 1997.
35. Chaby R, Caroff M. Lipopolysaccharides of Bordetella pertusis endotoxin. In: Wardlaw AC, Parton P, editors. Pathogenesis and immunity in Pertussis. New York: John Wiley & Sons; 1988. p. 247-71.
36. PPO 4 .09.037.03. Procedimiento establecido para determinar la inmunogenicidad del PRP-T en el CIGB. La Habana: CIGB; 2013.
Received in September, 2015.
Accepted in December, 2015.
Néstor Expósito. Dirección de Desarrollo Tecnológico, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba. E-mail: nestor.exposito@cigb.edu.cu.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}