










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.33 no.3 La Habana July.Sept. 2016
TECNIQUE
Improvements in the QUALITATIVE HCV UMELOSA test to detect the RNA of the hepatitis C virus
Mejoras al ensayo UMELOSA HCV CUALITATIVO para la detección del ARN del virus de la Hepatitis C
Yenitse Perea1, Anny Armas1, Yaimé González1, Juan E Figueredo1, Celia M Laza2, Boris E Acevedo3
1 Laboratorio de Biología Molecular, Subdirección de Inmunoquímica, Centro de Inmunoensayo, CIE. Calle 134 y Ave 25, Cubanacán, Playa, AP 6653, La Habana, Cuba.
2 Departamento Central de Matemática y Programación, Centro de Inmunoensayo, CIE, La Habana, Cuba.
3 Grupo de Negocios y Desarrollo de Proyectos. Centro de Ingeniería Genética y Biotecnología, CIGB. Ave 31 e/ 158 y 190, CP 11600, Cubanacán, Playa, La Habana, Cuba.
ABSTRACT
QUALITATIVE HCV UMELOSA is a test used since 2005 to confirm HCV infection and the follow-up of the antiviral therapy in patients infected with the hepatitis C virus (HCV). The drawbacks of the assay are the need for two rounds of the polymerase chain reaction and that it does not detect amplification inhibitions, leading to false positive and negative results, respectively. Therefore, the general objective of this study was to modify the QUALITATIVE HCV UMELOSA test without affecting its analytical characteristics. RNA extraction, amplification and detection stages of HCV were standardized. An internal standard was incorporated in the extraction step and a specific primer was introduced for its detection, to detect the presence of PCR inhibitors. Appropriate primers were introduced that made it possible to eliminate the nested PCR and therefore decrease the probability of obtaining false positive results. Finally, the analytical characteristics of the new test gave good clinical and analytical results and it was 100 % consistent with the reference kit. The new standardized trial showed a sensitivity of 72.31 IU/mL, which was higher than that of QUALITATIVE HCV UMELOSA (200 IU/mL), thereby enabling its use as a confirmatory assay in therapeutic follow-up and for the screening of plasma mixtures for hemoderivative production. The modifications made on QUALITATIVE HCV UMELOSA make it socially possible to offer better care for the patient with economic benefits because of the reduction of the production cost of the kit and the cost of the Cuban health system.
Keywords: RNA, HCV, Qualitative HVC UMELOSA, PCR, assay sensitivity, internal standard, inhibitors.
RESUMEN
El ensayo UMELOSA HCV CUALITATIVO se emplea desde el año 2005 en la confirmación de la infección por el virus de la hepatitis C (VHC) y el seguimiento de la terapia antiviral. Tiene como desventajas el empleo de dos rondas de reacción en cadena de la polimerasa (PCR) y la no detección de inhibiciones de la amplificación, con resultados falsos positivos y negativos. Por ello, el objetivo de este trabajo fue la modificación del ensayo UMELOSA HCV CU-ALITATIVO, para incrementar su sensibilidad y sin afectar sus características analíticas. Se estandarizaron las etapas de extracción del ARN, su amplificación y la detección del VHC, se añadió un estándar interno en la extracción y una sonda específica para su detección, para detectar la presencia de inhibidores de la PCR. Además, se introdujeron cebadores específicos para disminuir la probabilidad de obtener resultados falsos positivos, y se evaluaron las características analíticas del ensayo modificado. Este mostró buena especificidad clínica, analítica y una concordancia del 100 % con el estuche de referencia, y una sensibilidad de 72.31 UI/mL, superior a la del UMELOSA HCV CUALITATIVO (200 UI/mL). Esto permite su uso como prueba confirmatoria en el seguimiento terapéutico, y para la pesquisa de mezclas de plasma para la producción de hemoderivados. Además, desde el punto de vista social, este ensayo permite una mejor atención al paciente y aporta un beneficio económico al disminuir el costo de producción del estuche y los costos para el sistema de salud cubano.
Palabras clave: RNA, VHC, UMELOSA VHC cualitativo, PCR, sensibilidad del ensayo, estándar interno, inhibidores.
INTRODUCTION
The prevalence of the hepatitis C virus (HCV) infection was estimated by the World Health Organization (WHO) to be 3 %, which represents approximately 185 million persons infected in the world [1]. The infection by this virus may be expressed as acute hepatitis, frequently asymptomatic, which can lead to a chronic infection in 80 % of the cases [2]. Patients with chronic infection normally are asymptomatic for long periods of time. Approximately 20 % of these individuals develop hepatic cirrhosis after a 20 year evolution. Patients with cirrhosis, besides the risk of liver failure and the need of a transplant, have a highly significant risk of developing hepatocellular carcinoma.
At present, the infection by HCV is the main cause of liver transplant and of hepatocarcinoma in the western world, representing the main cause of hepatic morbidity and mortality [3]. In Cuba, approximately 1.38 % of the blood donor population has positive antibodies to HCV [4]. Liver cirrhosis is the tenth cause of death in women and the eleventh in men; in these, HCV has a leading role [5]. It is therefore considered that HCV is a serious health problem in our country.
The selected diagnostic method is the serological determination of anti-hepatitis C antibodies, which is highly sensitive [6]. Positive serological results should be confirmed with an additional test, which is now the most frequently used technique, i.e. the detection of viral ribonucleic acid (RNA) in circulation using Reverse Transcriptase Polymerase Chain Reaction (RT-PCR).
QUALITATIVE HCV UMELOSA has been produced since 2005 (Center of Immunoassay, CIE, Cuba), which is the first Cuban molecular diagnosis test for the confirmation of HCV infection. Its advantages include the detection of the infection in the window period between the infection and seroconversion, enabling the diagnosis of immunodepressed persons, and the confirmation of the infectious cases resolved (seropositive patients without viral replication) [7].
MATERIALS AND METHODS
Extraction of RNA from the HCV using QUALITATIVE HCV UMELOSA
The extraction of the RNA by QUALITATIVE HCV UMELOSA from the human serum or plasma was made through modifications of the method described by Chomczynski and Sacchi in 1987 [8]. It was started by adding 500 μL of the lysis buffer (0.3-0.6 mol/L ammonium tiocyanate (BDH, Poole, England); 0.6-1.0 mol/L guanidine tiocyanate (Sigma, Saint Louis, USA); phenol (v/v) 35-40 % (Merck, Darmstadt, Germany); 5-8 % v/v Glycerol (Sigma, Saint Louis, USA); 0.1-0.3 mol/L sodium acetate (Sigma, Saint Louis, USA); pH of 4 to 5 adjusted with glacial acetic acid (Merck, Darmstadt, Germany) and 0.05-0.1 mg/mL yeast RNA (BDH, Poole, England), water treated with diethylpyrocarbonate and sterilized (Sigma, Saint Louis, USA)) [9] to 150 μL of human serum or plasma.
After vigorously mixing, 100 μL of chloroform were added to each tube with shaking the mixture for 15 s, and centrifuged at 12 000 g for 15 min, at 23 ºC. Then, the supernatant was collected and added to another tube containing isopropanol (Merck, Germany) in a proportion of 1:1. After homogenization by inversion, another centrifugation was carried out at 12000 g for 10 min and at 4 ºC, giving rise to the RNA precipitate. A washing step of the precipitate was made with 75 % ethanol (Merck, Germany) and finally the precipitate was dried at 22-25 ºC and resuspended in 20 μL of ribonuclease-free water.
Modifications made in the extraction of the RNA
Selection of the sample volume
To select the volume of extraction of the serum or human plasma, we evaluated a negative control and four positive samples at low concentrations of RNA of the HCV (2000, 500, 250 and 100 IU/mL), prepared from a secondary standard with 3.52 × 105 IU/mL. The RNA was extracted from two replicates of the negative control and from four replicates of each one of the positive samples, starting from different sample volumes: 150, 200 and 500 μL.
Establishing the dilution of the internal standard (IS) in the RT-PCR assay
The RNA was isolated using four replicates of two negative controls and two replicates of two secondary standards: UMECONT-1 and UMECONT-2, with high viral loads, quantified by the Cobas TaqMan HCV test v1.0 (Roche Diagnostics, Germany), resulting in 1.43 × 106 IU/mL and 3.52 × 105 IU/mL, respectively. A lot of the IS transcript (T-IS) was used as the internal quantification standard; it contained a sequence of the p64K protein from Neisseria meningitidis and from the same primer binding sites as the sequence of RNA from HCV, which was added to the lysis buffer in the dilutions of 1:4 × 106, 1:4 × 107 and 1:4 × 108 to determine the competing dilution.
Inverse-amplification transcription of the competitive RT-PCR
The synthesis of the complementing deoxyribonucleic acid through inverse transcription and its amplification by the polymerase chain reaction was made in the same vial using 25 μL of a reaction mixture and 5 μL of the isolated RNA, which had been resuspended in water free from RNases. The reaction mixture was standardized for a similar trial using the same primers [10], containing the buffer Go Taq; MgCl2 at 1.5 mM, 250 μM of each dNTP; 3.6 U of the inverse transcriptase AMV-RT, 7.5 U of the ribunuclease inhibitor RNasin and 1.05 U of the polymerase DNA Go Taq Flexi (all of which were from Promega, USA). The program for the amplification was the following: firstly, a step at 45 °C for 30 min, followed by a denaturion step at 95 °C for 3 min, 50 thermal cycles (95 °C, 30 s; 67 °C, 30 s; 72 °C, 30 s), and finally an extension step at 72 °C for 7 min. The RT-PCR was carried out in a thermo-cycler with a gradient (Eppendorf AG 22331, Germany) [11].
Modifications in the amplification step
Selection of the volume of the RT-PCR mixture and of the volume of purified RNA
The RNA was isolated using four replicates of the negative control and six replicates of four positive samples with low concentrations of RNA from the HCV (2000, 500, 250 and 100 IU/mL) prepared from the UMECONT-2. The extraction of the RNA from the HCV was made from a volume of the sample of human serum or plasma giving the best result in the trial, and in the RT-PCR the following combinations of the mixture of RT-PCR and purified RNA were assessed: (25 μL of the mixture RT-PCR and 5 μL of RNA; 20 μL of the mixture RT-PCR and 10 μL of RNA and 15 μL of the mixture of RT-PCR and 15 μL of RNA).
Hybridization and detection of the amplified product through chemical denaturation
The qualitative detection of the amplified product was carried out through hybridization in the UMELOSA® (ultra micro-analytic enzyme-linked oligonucleotide-sorbent assay) format. The amplicons having the sequence of HCV and the internal standard (IS) were captured in duplicates on alternative strips of polystyrene ultramicroplates (Greiner Bio-One, Germany) coated with the primers: Q5 (5´NH2-TCG TGC AGC CTC CAG GAC CCC CCC TCC CGG 3´) and S1 (5´NH2-TAG TTG AAT TGA AAG TGC CCG ACA T 3´), respectively. Before application in the wells of the plates, we added the denaturalization solution containing NaOH in each test tube and after incubating for 10 min at room temperature (22-25 ºC) the denaturalized amplified products were placed in the respective wells in the ultramicro-plate.
Later, the neutralization-hybridization solution formed by HCl, sodium salts and SDS [12] was applied and the plate was incubated for 1 h at 37 ºC in a humid chamber. After that time, the microplate was washed and the conjugate of alkaline Streptavidine/ Phosphatase, diluted at 1:500 000 was added; it was incubated at 37 ºC in a humid chamber for 30 min and after washing the plate, the substrate 4-Metilumbeliferil phosphate was added. The fluorescence signals corresponding to the hydrolysis of the substrate by the conjugate bond to the biotinilated amplicons were detected by the plate reader PR-621 (TecnoSuma, CIE, Cuba).
Hybridization and detection of the amplified product using thermal denaturation
The qualitative detection of the amplified product using thermal denaturation was carried out according to the above description, but substituting the chemical denaturalization step. It was then added the neutralization-hybridization solution for the incubation of the amplified products at 95 °C for 10 min [13, 14] and the later inclusion of a combined hybridization solution with sterile purified water at the proportion of 1:1.
Modifications in hybridization and detection
Selection of the denaturation method of the amplified products
The RNA was isolated, starting from the four negative controls and the twenty replicates of a sample with 250 IU/mL of RNA of the HCV prepared from UMECONT-2. The extraction was made from 500 μL of the volume of the serum or human plasma sample and the amplification was done by RT-PCR according to the method described above, using 15 μL of the volume of the mixture of RT-PCR and 15 μL of the volume of isolated RNA. The hybridization and detection were carried out with chemical denaturation of the amplified products, with two replicates of the negative controls and ten replicates of a sample with 250 IU/mL of RNA of the HCV. Meanwhile, the hybridization and detection of the amplified products of the rest of the negative controls and the replicates of the sample with 250 IU/mL of RNA of the HCV was made using thermal denaturalization.
Evaluation of the analytical characteristics of the modified assay
For the evaluation of the analytical characteristics, HCV RNA was extracted from 500 μL of the volume of the sample of human serum or plasma and later amplified by RT-PCR using the combination of 15 μL of the sample of RT-PCR and 15 μL of purified RNA. The hybridization and detection of the amplified products was carried out using thermal denaturalization.
Clinical specificity
In order to validate clinical specificity of the new trial, we evaluated 146 samples that were seronegative to UMELISA HCV and negative to QUALITATIVE UMELOSA HCV. Clinical specificity was calculated using the following equation:
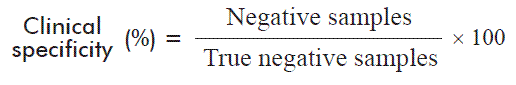
Where the true negative samples are the negative samples plus the false negative samples, established by a reference method.
Analytical specificity
A test was made on five positive samples to the human immunodeficiency virus type 1 (HIV-1) and 2 (HIV- 2), five positive samples to Chagas disease and five positive samples to the human hepatitis B virus.
Evaluation of sensitivity using an international standard of HCV RNA
The sensitivity of the modified trial was evaluated using the international standard (IS) (06/100), genotype 1a, with the concentration of 154 881 IU/mL of RNA of the HCV, from which the following concentrations were prepared: 200, 100, 50, 25, 12.5 and 6.25 IU/mL. The replicates were tested on different days and by different operators. The RNA was isolated with 24 replicates of each one of these concentrations. In this study, a sample was regarded as positive when the fluorescence value was above the mean of the negative control plus three times its standard deviation. The PROBIT analysis [15] was carried out to determine the detection limit of the assay [16].
Evaluation of genotypic specificity
The RNA was isolated from four replicates of genotypes 1 to 6 of the Genotyping Panel 08/264 of NIBSC, with a concentration range of 500 to 1500 IU/mL.
Robustness
Alternately, 22 samples (11 negative and 11 positive samples) to the RNA of the HCV were evaluated to test the capacity of the test in preventing crossed contaminations. The positive samples with a concentration of 10 000 IU/mL were prepared from a secondary standard.
Evaluation of the capacity of the trial to detect inhibitory samples of RT-PCR
Several inhibition tests were designed, taking into account reports from the literature, to demonstrate that the modified test is able to detect the presence of inhibitory substances of the PCR. Several types of inhibitors were tested, such as: organic solvents (acetone, ethanol and isopropanol), salts (sodium acetate and sodium chloride) and hemolyzed blood, by freezing-thawing. In each test we included a negative control and a positive control to 7500 IU/mL of HCV RNA. Each inhibition variant was tested by triplicate. Total inhibition was considered when the fluorescence signals of HCV and IS were below 20 fluorescence units. The extraction of the RNA of the HCV was made from 500 μL of the sample of serum or human plasma with the following features: volumes of 0.7; 1.0; 1.5; 3.0 and 5.0 μL of the organic solvents: acetone, ethanol and isopropanol, were added to the 20 μL of water in which the extracted RNA was resuspended. The saline solutions of sodium acetate at 0.006; 0.01; 0.02 and 0.04 mol/L and of sodium chloride at 0.03; 0.06 and 0.12 mol/L were evaluated by resuspending the RNA in 20 μL of these solutions instead of using water free from ribonucleases. The hemolyzed blood used in this study was obtained by freezing-thawing of a sample of human blood that was negative to HCV, HIV and HBV. It was used as a diluent to prepare a sample that is positive to 7500 IU/mL of RNA of the HCV.
Comparison between modified QUALITATIVE HCV UMELOSA and QUALITATIVE HCV UMELOSA
RNA was extracted from 483 samples: 146 negative samples and 337 positive samples through the QUALITATIVE HCV UMELOSA using 150 μL and the amplification was carried out by RT-PCR as described in the QUALITATIVE HCV UMELOSA kit, v. 2, 2012) [12], using 25 μL of a reaction mixture of RT-PCR and 5 μL of the purified RNA. The extraction of the HCV RNA using the new variant of the QUALITATIVE HCV UMELOSA test was made with the same samples and using 500 μL of the volume of the human serum or plasma sample. The amplification through RT-PCR was carried out using 15 μL pf the RT-PCR mixture and 15 μL of purified RNA. The hybridization and detection of the amplified products was carried out using thermal denaturalization. The percentage of coincidence between both tests was also calculated.
Statistical analysis
In the experiment made to determine the sensitivity of the modified test with the international standard (06- 100), the statistical package MINITAB Release 14 (Minitab Inc. Pennsylvania, USA.) and the PROBIT analysis [15] were used to determine the detection limit at 95 % and its confidence interval [15].
In the experiment to select between the thermal and chemical denaturation, the means were compared using the Student’s t test. The differences observed between the means were estimated at a 95 % confidence interval (CI), with a significance level (p) of 0.05, all these determined with the statistical package STATISTICA 6.0 (StatSoft Inc., 2001, Oklahoma, USA).
RESULTS AND DISCUSSION
Modifications in the RNA extraction
Selection of the sample volume
As observed in table 1, the volume of the sample for which the best percentages of positivity were obtained for all concentrations of RNA of the HCV evaluated was 500 μL. Both the volume of the sample used in the QUALITATIVE HCV UMELOSA test (150 μL) and 200 μL of the sample did not enable the detection of the lowest concentration of RNA of the HCV evaluated (100 IU/mL), which coincides with the detection limit of the QUALITATIVE HCV UMELOSA test using a secondary standard [17]. In all cases, the replicates of the negative control gave signs of fluorescence of the negative HCV.
Establishing the competition dilution of the IS in the RT-PCR assay
As shown in table 2, the optimum dilution of the T-IS as an internal standard was 1:4×106 since with this standard the highest fluorescence signals were obtained for the internal standard of the negative controls. This makes it possible for the trial to readily detect the presence of inhibitions before which the fluorescence signal corresponding to the internal standard would fall.
Furthermore, with the dilution of 1:4×106 of the T-IS as an internal standard, the high fluorescence signals of the HCV were obtained for the positive controls, which are values of interest for both controls (data not shown).
A dilution of less than T-IS was not evaluated because higher concentrations of the internal standard favor the fluorescence signal of the IS and significantly decrease the fluorescence signal of the HCV for positive samples.
Modifications in the amplification
Selection of the volume of the RT-PCR mixture and the volume of the purified RNA
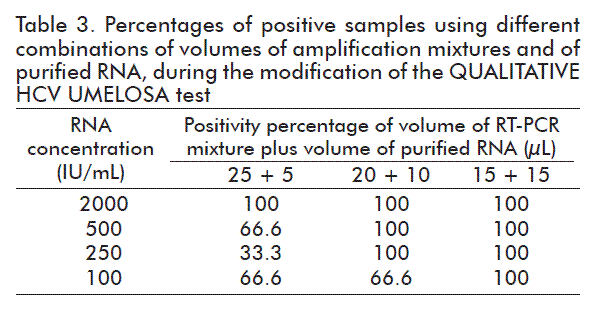
As observed in table 3, the best combination was that of 15 μL of the mixture of RT-PCR and 15 μL of purified RNA since, with this combination, 100 % positivity was obtained for all concentrations evaluated.
Modifications in the hybridization and detection
Selection of the denaturation method of the amplified products
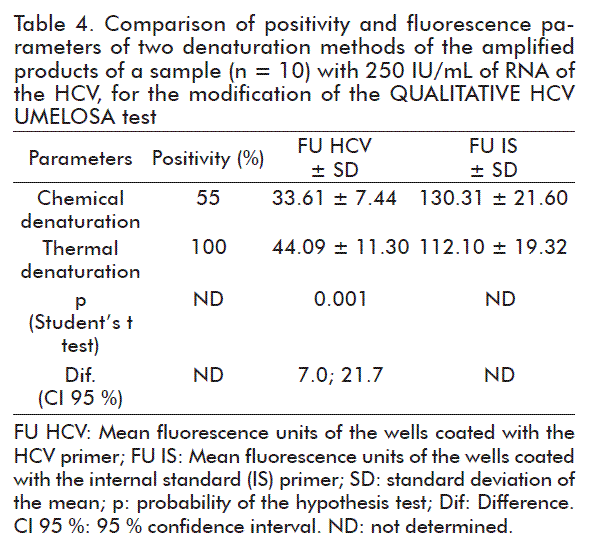
The value of the mean fluorescence corresponding to the replicates of 250 IU/mL that are thermally denatured, was significantly higher than the one corresponding to chemically denatured replicates (p < 0.05) and the confidence interval for the difference of the means does not contain the zero value and at the same time it is positive (Table 4).
The best variant selected was thermal denaturalization, because it detected 100 % of the ten replicates of the sample with 250 IU/mL of RNA of the HCV as observed in table 4. Furthermore, this method has the advantage of not requiring the addition of denaturation and neutralization solutions, with which there is a saving in time, reagents and others.
Clinical specificity
The clinical specificity of a TAN (technical amplification of nuclear acids) test consists of its capacity to determine unequivocally the presence of a specific DNA or RNA sequence within a large variety of other sequences. It depends, first of all, on the selection of the primers for the detection of the amplified product and the conditions of the amplification and detection processes. It is also defined as a proportion of the negative samples (non-reactive) that are correctly identified by the test used.
All samples evaluated were negative according to the MODIFIED QUALITATIVE HCV UMELOSA, obtaining 100 % clinical specificity for the new test.
Analytical specificity
On studying the analytical specificity of the MODIFIED QUALITATIVE HCV UMELOSA, all samples gave negative results. This is possible because the specificity of the PCR is based on the correct binding of the primer to the DNA target and in the ability of the polymerase to appropriately replicate the selected region.
Evaluation of the sensitivity using an international HCV RNA standard
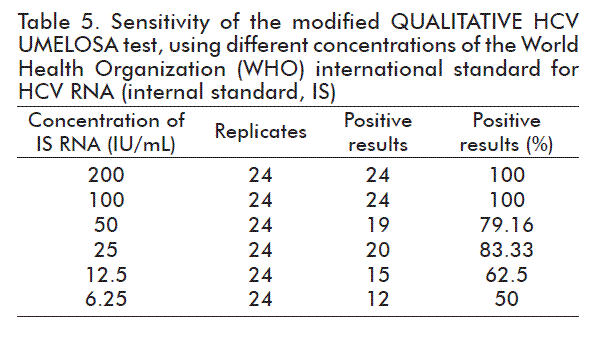
In all cases, the expected results were obtained for the positive control and the negative control included in each setting (data not shown). The results obtained for each concentration of the IS (International Standard) evaluated are shown on table 5.
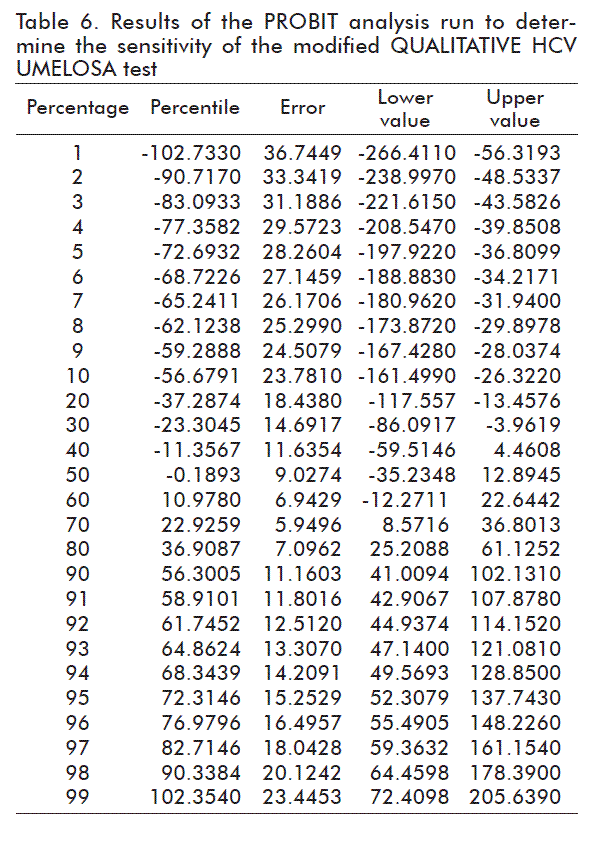
The PROBIT analysis showed that the 95 % detection limit was at 72.31 IU/mL, with a CI of 52.31 to 137.75 IU/mL (Table 6).
The sensitivity obtained with the IS (72.31 IU/mL) is better than that reported for the QUALITATIVE HCV UMELOSA with the IS (200 IU/mL) [17]. The differences may be due to the fact that both studies for sensitivity were carried out with different amounts of replicates, the international standards used do not have the same genotypes, or due to the changes made in the extraction, amplification and detection methods in the modified QUALITATIVE HCV UMELOSA.
FDA demands a sensitivity or limit of detection (LD) of 95 % of 5000 IU/mL [16] for the screening of the HCV in individual blood bags, while to evaluate mixtures of plasma in blood Banks or for the use of hemoderivatives in Europe, a minimum sensitivity of 100 IU/mL or less is required [18, 19]. The sensitivity of the modified test (72.31 IU/mL) complies with the requirements of international regulations for the screening of blood or plasma bags for hemoderivatives, as long as the number of bags to be mixed is regulated according to the sensitivity of the trial. In our case, the mixtures would be of 50 bags, in theory, but in practice it has been demonstrated that it is best to work with mixtures of 24 bags, thus minimizing the risk that a positive bag, with a very low concentration, may escape the screening [20]. On evaluating the IS (Table 5), it is observed that 100 IU/mL (n = 24) are detected 100 % of the times.
At present, the producers of qualitative clinical trials compete to reach lower detection limits. Examples of this are the tests produced by Roche Molecular Systems (Pleasanton, USA), the leader in this market being AMPLICOR® HCV Test v 2.0, having a LD at 95 % of 60 IU/mL in the serum and of 50 IU/mL in the plasma [21]; the COBAS AMPLICOR® HCV Test v 2.0, with 60 IU/mL in the serum and 50 IU/mL in the plasma [22], and the COBAS Ampliprep/Cobas Taqman HCV Test (although it has another type of assay, real time PCR, and it is designed specifically for screening in blood banks) with 28 IU/mL [23]. These very low detection levels are not only for the earlier detection of a patient during the window phase (the acute phase of the disease), detecting the patient during screening in blood Banks, therefore declining to zero the transmission by this route, or adequately monitoring the treatment. They are also established to assure a very profitable market which is almost exclusive for the large diagnostic agents producing companies in the world (Roche, Siemens and Abbott), which makes it unaffordable for a developing country to use commercial assays having this ‘quality’ for the free of charge diagnosis and follow-up of the treatment in its population.
Evaluation of genotypic specificity
With the modifications made on the QUALITATIVE HCV UMELOSA, all the genotypes from 1 to 6 from the Genotyping Panel 08/264 (NIBSC) with a concentration range of 500 to 1500 IU/mL (four replicates of each genotype were detected at all times (100 %) Genotyping panel 08/264 (NIBSC). These results are in agreement with those obtained in the genotypic sensitivity study made on the QUANTITATIVE HCV UMELOSA having a similar format to ours, in which the six genotypes of the Genotyping panel 02/202 were detected at 1000 IU/mL in 100 % of the tests [9].
Recent studies carried out on Cuban patients with chronic HCV show that the subtype 1b of HCV is prevalent in Cuba and the subtype 1a has a low prevalence [24]. In that study, based on the analysis of the sequence of the region coding for the capsid protein of HCV, the viral genotype was determined in 58 Cuban patients who were positive for HCV RNA. Here we obtained a prevalence of 94.8 % for subtype 1b and of 5.2 % for subtype 1a. This study also revealed the mutability of subtype 1b in Cuba, making it prevalent, since this allows for its fast propagation, producing a negative impact on antiviral therapy in our country [24]. In another study carried out by Roca et al., subtype 1b also had the highest prevalence in 33 Cuban patients studied, constituting 42.4 %; subtypes 1a and 2b showed moderate prevalence (9.09 %) and subtype 3a was found in 35 % of Cuban patients and it was always found as a co infection with subtype 1b [25]. In 2012, another study carried out in 79 Cuban patients with HCV was published; 10 of them (12 %) had genotype 1a and 69 had genotype 1b (82 %). Five patients (6 %) had combined infections, among which two (2 %) had genotype 1a/3a and three had genotype 1b/3a, 1b/4a and 1a/1b/3a, respectively [26].
Robustness
The robustness of an analytical procedure offers a measure of its confidence for routine use. There was no cross contamination between the positive samples with high viral loads and the analyzed negative samples. All negative samples and the negative control of the trial were negative in the test, while the positive sample and the positive control were positive. Similar results were obtained with the QUALITATIVE HCV UMELOSA when evaluating fourteen alternate samples, seven were positive with high viral load and seven were negative, to test the capacity of the trial avoiding crossed contamination [17].
Evaluation of the capacity of the test to detect samples that inhibit RT-PCR
The samples can contain inhibitors such as hemoglobin, cloth, tissues and soils. Other important sources of inhibitors are the materials and reagents that get into contact with the sample during its processing. These include excess KCl, NaCl and other salts, and organic solvents such as ethanol, phenol and isopropanol.
Phenol at a concentration higher than 0.2 % inhibits the PCR reaction, and ethanol, isopropanol and acetone at concentrations higher than 1 %. Certain saline solutions such as sodium acetate at above 5 mmol/L and sodium chloride at above 25 mmol/L are also inhibitory [27].
As shown in table 7, when 1 μL of isopropanol and 5 μL of acetone and ethanol were added, a total inhibition of the PCR was produced (threshold set to less than 20 FU). Our test was more sensitive to the presence of small volumes of residual isopropanol. When using saline solutions, sodium acetate produced complete inhibition at 0.04 mol/L and sodium chloride at 0.12 mol/L. The use of hemolyzed blood by freezing-thawing also produced the complete inhibition of PCR (Table 7).
It is very important to detect inhibition to avoid false negative results. For this, we introduced an IS in the lysis buffer, to detect the inhibitions due to the fall of the fluorescence signals both of HCV and IS.
PCR is extremely sensitive and reproducible when analyzing samples of pure DNA. Nevertheless, the efficiency of amplification and the detection limit are frequently altered by the interference of different molecules with DNA polymerase, or with the nucleotides and nucleic acids.
The interactions between the proteins and DNA polymerase are the main cause of PCR inhibitions. The human blood contains more proteins than DNA; many of these proteins interfere with the polymerization process that occurs in the PCR. An example of this is the Heme group, which acts as a human blood inhibitor because it frees the iron ions affecting the ionic balance and altering polymerase activity [28]. Isopropanol, ethanol and acetone produce the precipitation of nucleic acid, which does not favor its replication because the probability of interaction of the replicative machinery with nucleic acids is affected. The K+ ions are frequently used in the PCR regulating solutions to ensure the appropriate ionic environment. Changing the K+ content or introducing other ions such as those of Na+ may produce inhibition [28].
Comparison of modified QUALITATIVE HCV UMELOSA and QUALITATIVE HCV UMELOSA
A complete agreement (100 %) was obtained between the results of both tests. A similar result was achieved on comparing QUALITATIVE HCV UMELOSA with the AMPLICOR HCV Test, v 2.0, which showed a 100 % agreement between both tests [17].
The new standardized test (modified QUALITATIVE HCV UMELOSA) was able to reduce in 3 h the assay period compared to QUALITATIVE HCV UMELOSA, because it reduces hybridization time to 1 h and uses only one round of PCR. Modified QUALITATIVE HCV UMELOSA showed better sensitivity than QUALITATIVE HCV UMELOSA, while preserving its analytical characteristics. Furthermore, it uses lower amounts of reagents than the current test, thereby reducing its production cost. It also has the advantage of detecting samples that inhibit PCR, thereby preventing false negative results, and, on the other hand, the probability of obtaining false positives is decreased on eliminating the second amplification round, making it possible for the results to be issued in a more reliable and rapid manner. Both changes, from the social viewpoint, offer a better care for the patient and economic benefits because they decrease the production cost of the kit and the costs to the Cuban health system.
In summary, with this optimized test, under sustainable production, which is available for our national health programs, a very valuable working tool is available that has the sensitivity and quality to comply with the internationally established requirements for TAN assays.
REFERENCES
1. Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333-42.
2. Colquillo B, Sanchez R, Terceros P. Nuevas estrategias para el diagnóstico y seguimiento de la Hepatitis C. Evaluación de las técnicas de laboratorio RT PCR y EIA. Biofarbo 2009:17(2):15-22.
3. Ly KN, Xing J, Klevens RM, Jiles RB, Ward JW, Holmberg SD. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann Intern Med. 2012;156(4):271-8.
4. TecnoSUMA. Control de enfermedades infecciosas. 2015 [cited 2015 Dec 12]. Disponible en http://www.tecnosuma.com/programasnacionales.
5. Ministerio de Salud Pública. Anuario Estadístico de Salud 2012. La Habana: Dirección Nacional de Registros Médicos y Estadísticas de Salud, MINSAP; 2013.
6. Hernández J. Evolución del virus de la Hepatitis C en muestras Hospitalarias de la Comunidad Valenciana (Tesis Doctoral): Valencia: Servei de Publicaciones, Universidad de Valencia; 2005.
7. Gonzalez-Perez I, Gonzalez Gonzalez YJ, Vina-Rodriguez A, Armas Cayarga A, Solis RL. The usefulness of Umelosa hepatitis C virus qualitative kit as supplemental test for confirmation of hepatitis C virus infection. Rev Soc Bras Med Trop. 2004;37(1):25-7.
8. Gonzalez-Perez I, Armas Cayarga A, Garcia de la Rosa I, Josefina Gonzalez Gonzalez Y. Homemade viral RNA isolation protocol using silica columns: a comparison of four protocols. Anal Biochem. 2007;360(1):148-50.
9. Gonzalez-Perez I, Cayarga AA, Hernandez YP, de la Rosa IG, Gonzalez YJ, Leon CS, et al. Long-term conservation of HCV RNA at 4 degrees C using a new RNA stabilizing solution. J Virol Methods. 2010;168(1-2):207-11.
10. TecnoSUMA. Instructivo del estuche QUANTITATIVE HCV UMELOSA. Edición 2. La Habana: TecnoSUMA; 2010.
11. Perea Y, Armas A, González YJ, Laza CM. Stability of standard curves of hepatitis C virus transcripts used for viral quantification. Biotecnol Apl. 2009;26(3):222-5.
12. TecnoSUMA. Instructivo del estuche QUALITATIVE HCV UMELOSA. Edición 2. La Habana: TecnoSUMA; 2012.
13. Máster Diagnóstica. Instructivo del estuche HPV Direct Flow Chip Kit, v 1.0. Granada: Máster Diagnóstica; 2014.
14. Esteban J, Alonso-Rodriguez N, del- Prado G, Ortiz-Perez A, Molina-Manso D, Cordero-Ampuero J, et al. PCR-hybridization after sonication improves diagnosis of implant-related infection. Acta Orthop. 2012;83(3):299-304.
15. Finney DJ. PROBIT analysis; parallel line analysis. Tercera edición. Cambridge: Cambridge University Press; 1971.
16. Jose M, Gajardo R, Jorquera JI. Stability of HCV, HIV-1 and HBV nucleic acids in plasma samples under long-term storage. Biologicals. 2005;33(1):9-16.
17. Gonzalez-Perez I, Gonzalez Gonzalez YJ, Armas Cayarga A, Vina-Rodriguez A, Medina Concepcion A, Trujillo Pelegrin N, et al. Validation of a nested PCR assay UMELOSA HCV CUALITATIVO for the detection of Hepatitis C virus. Biologicals. 2003;31(1):55-61.
18. Tabor E. Workshop on implementation of Nucleic Acid Testing. December 14, 1999. Bethesda: National Institute of Health, Bethesda; 1999.
19. Saldanha J, Heath A, Lelie N, Pisani G, Nubling M, Yu M. Calibration of HCV working reagents for NAT assays against the HCV international standard. The Collaborative Study Group. Vox Sang. 2000;78(4):217-24.
20. Fryer JF, Minor PD. Standardisation of nucleic acid amplification assays used in clinical diagnostics: a report of the first meeting of the SoGAT Clinical Diagnostics Working Group. J Clin Virol. 2009;44(2):103-5.
21. Martins PP, Lampe E, Lewis-Ximenez LL, de Souza PS, Fernandes CA, Villar LM. Performance of molecular methods for hepatitis C virus diagnosis: usefulness among chronic cases and during the course of infection. Clin Lab. 2013;59(9-10):1031-9.
22. Cabezas-Fernandez M, Cabeza-Barrera M. Introduction of an automated system for the diagnosis and quantification of hepatitis B and hepatitis C viruses. Open Virol J. 2012;6:122-34.
23. Vermehren J, Yu ML, Monto A, Yao JD, Anderson C, Bertuzis R, et al. Multi-center evaluation of the Abbott RealTime HCV assay for monitoring patients undergoing antiviral therapy for chronic hepatitis C. J Clin Virol. 2011;52(2):133-7.
24. Gonzalez-Horta EE, Marante J, Amador-Canizares Y, Alvarez-Lajonchere L, Guerra I, Martinez-Donato G, et al. Analysis of hepatitis C virus core encoding sequences in chronically infected patients reveals mutability, predominance, genetic history and potential impact on therapy of Cuban genotype 1b isolates. Eur Rev Med Pharmacol Sci. 2011;15(11):1320-7.
25. Roca J, Ahmad M, Panda SK, Padrón G, Jameel S. Hepatitis C virus genotyping in developing countries: Results from Cuba, India and Turkey. Biotecnol Apl. 1999;16(2):88-92.
26. Rodriguez Lay Lde L, Villalba MC, Corredor MB, Frometa SS, Hernandez JM, Carrera SD, et al. HCV genotype determination in monoinfected and HIV co-infected patients in Cuba. Trans R Soc Trop Med Hyg. 2012;106(12):711-7.
27. Somma M. Sesión 4. Extracción y purificación de ADN. In: Querci M, Jermini M, Van den Eede G (eds.). Análisis de la presencia de microorganismos genéticamente modificados en muestras de alimentos. Ispra: Institute for Health and Consumer Protection; 2002; p. 1-18.
28. Hedman J, Radstrom P. Overcoming inhibition in real-time diagnostic PCR. Methods Mol Biol. 2013;943:17-48.
Received in February, 2016.
Accepted in December, 2016.
Yenitse Perea. Laboratorio de Biología Molecular, Subdirección de Inmunoquímica, Centro de Inmunoensayo, CIE. Calle 134 y Ave 25, Cubanacán, Playa, AP 6653, La Habana, Cuba. E-mail: yenitse.perea@cie.cu.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}