










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.34 no.1 La Habana Jan.-Mar. 2017
TECHNIQUE
Evaluation of viral RNA thermostability stored as dry pellet at room temperature
Evaluación de la termoestabilidad del ARN viral almacenado como pellet seco a temperatura ambiente
Damarys Relova, Lester J Pérez, Liani Coronado, Yoandry Hinojosa, Liliam Ríos, Ana María Acevedo, María Teresa Frías, Carmen L Perera
Departamento de Virología, Centro Nacional de Sanidad Agropecuaria, CENSA. Apartado 10, San José de las Lajas, Mayabeque, Cuba.
ABSTRACT
There is now widespread use of molecular tools for diagnosis and study of viral diseases. For this, it is essential to preserve the quality of genetic material while it is stored. Most laboratories store nucleic acid at ultra-low temperatures. However, there is a growing tendency on the search for new alternatives that allow storage of nucleic acid at room temperature. In this work the thermal stability of viral RNA retained as a dry pellet inside a sealed polyethylene tube and stored at 4, 20 and 37 ºC for a period of two months was studied. The study was conducted through periodic quantification of the concentration of total RNA of classical swine fever virus (CSFV) and avian influenza virus (AIV) using the rRT-qPCR. The results showed the efficacy of preservation method used in this experiment. In each time evaluated it was possible to quantify the amplification of the 5’UTR and NS5B regions of CSFV and M and HA genes of avian influenza virus, regardless of the temperature at which the RNA samples were stored. It showed that viral RNA storage as dry pellet retained within a hermetically sealed polyethylene tube is an effective way to preserve the stability of the viral RNA for long periods at room temperature.
Keywords: viral RNA, dry pellet, thermostability, storage, rRT-qPCR.
RESUMEN
Dada la gama de herramientas moleculares disponibles para el diagnóstico y estudio de las enfermedades virales, es esencial preservar la calidad del material genético durante su almacenamiento. La mayor parte de los laboratorios almacenan las muestras de ácidos nucleicos a temperaturas ultrabajas. Sin embargo, existe una tendencia creciente hacia la búsqueda de nuevas alternativas que permitan el almacenamiento a temperatura ambiente. En este trabajo se estudió la termoestabilidad del ARN viral conservado en forma de pellet seco, en viales de polietileno herméticamente cerrados y almacenados a 4, 20 o 37 ºC durante dos meses. Las muestras se analizaron periódicamente mediante la cuantificación por rRT-qPCR de la concentración del ARN total del virus de la peste porcina clásica (VPPC) y el virus de la influenza aviar (VIA). El método de conservación ensayado fue eficaz a las temperaturas ensayadas. Fue posible cuantificar las muestras de VPPC y de VIA mediante la amplificación de las regiones 5’UTR y NS5B, y de los genes M y HA, respectivamente, con independencia de la temperatura de almacenamiento. Se demostró que el almacenamiento de la ARN viral conservado en forma de precipitado seco en viales de polietileno herméticamente cerrados es una forma efectiva para la preservación de la estabilidad del ARN viral durante largos períodos a temperatura ambiente.
Palabras clave: ARN viral, termoestabilidad, precipitado seco, almacenamiento, rRT-qPCR.
INTRODUCTION
With the increasing use of molecular techniques for the diagnosis and study of diseases, many laboratories have developed different strategies for the preservation and storage of genetic material without affecting its quality [1]. Regardless of the number and intrinsic differences of the types of samples from which the nucleic acids are obtained, their quality will depend on several factors such as: efficiency of the extraction method used, the type of matrix in which it they are stored, purity, ionic strengths, the quality of the container material, exposure to UV light, humidity and temperature range, as well as the time in which the sample is stored and its exposure to multiple freeze-thaw cycles [2].
The DNA molecule is very stable unlike RNA, which is highly vulnerable to degradation under extreme conditions, such as storage for long periods of time or at high temperatures [3]. RNA in aqueous environment can be degraded by the spontaneous cleavage of the phosphodiester bonds as a result of the acid or basic hydrolysis of the 2´-OH group of the phosphorus atom [4]. In addition, it is very sensitive to oxidation by reactive oxygen species, so it is also necessary to protect it from the oxygen contained in the atmosphere [5]. Oxidation could also be a result of ozone attack, an atmospheric pollutant that reacts rapidly with RNA, either in solution or in solid state [6].
For several years the most effective and least expensive ways of storing genetic material have been studied. The most widely used alternative for the preservation of nucleic acids for long periods of time has been aqueous matrices at freezing temperatures of –20 and –80 °C. Another widely used method has been the preservation and storage of nucleic acids in buffer solutions where they can keep their stability even at room temperature, but only for short periods of time [7, 8]. In recent years, researchers have paid special attention to the study of long-term storage systems for dehydrated RNA at room temperature [1, 9].
On the other hand, most of the diseases that have caused devastating losses in the agricultural sector have been caused by viruses, the most significant ones caused by RNA viruses such as: avian influenza virus (AIV), foot-and-mouth disease virus (FMDV), African swine fever virus (ASFV) and classical swine fever virus (CSFV), among others. The rapid and accurate confirmatory diagnosis of suspected outbreaks of these diseases is vital to take timely zoo-sanitary measures [10-12]. That is why the polymerase chain reaction (PCR) technique has become the diagnostic tool of choice for these entities, which makes it essential to guarantee the non-degradation of the RNA sample used for this diagnosis [13-18].
Many of these viral RNA samples are stored for long periods of time for use in subsequent epidemiological studies. Hence, it is necessary to evaluate efficient storage methods that ensure the preservation of viral RNA stability. Therefore, this work was aimed to evaluate the viral RNA thermostability stored as dry pellet at room temperature.
MATERIALS AND METHODS
Selection of working samples
For this study, RNAs of the AIV and of the CSFV were taken as experimental models. RNA samples were obtained from the hemagglutinin antigen of AIV type A subtype H5N1 (A/chicken/Egypt/0870-NLQP / 2008), kindly donated by the Institute of Virology of Padova, Italy, and the Cuban CSFV isolate, ‘Pinar del Río’ (CSF1058), from the microbial collection of the National Center for Agricultural and Livestock Health (CENSA), Cuba.
RNA extraction
Total RNA extraction was performed from 140 μL of each sample, using the commercial kit QIAamp Viral RNA Mini Kit (Qiagen GmbH, Germany), according to the manufacturer’s instructions.
RNA samples processing and storage
The isolated RNAs were distributed in 20 μL aliquots in 200-μL polypropylene tubes (Eppendorf AG, Hamburg, Germany) and to each tube was added 53.6 μL of precipitation solution (20 mg/mL Glycogen, 7.4 M Ammonium Acetate in absolute Ethanol). They were centrifuged at 12 000 g for 10 min, the supernatant was discarded and the RNA pellets were allowed to dry at room temperature until total dehydration.
The samples, once dehydrated, were tightly closed in the polypropylene tubes containing them and stored at three independent temperature conditions: 4, 20 or 37 ºC. The efficiency of the different conservation methods was evaluated periodically for two months. At each time point, three replicates per sample were rehydrated with nuclease-free water and analyzed by real-time quantitative RT-PCR (rRT-qPCR).
Complementary DNA synthesis
The complementary DNA (cDNA) was synthesized by using theMoloney-Murine leukemia virus reverse transcriptase enzyme (M-MLV RT) (Promega, Madison, WI, USA). For this purpose, a mixture of 7.7 μL of nuclease-free water (Promega, Madison, WI, USA), 1 μL of random primers (Promega, Madison, WI, USA; 50 ng/ μL), 1 μL DNTP (10 mM), 4 μL of the 5× reaction buffer, 0.5 μL of ribonucleases inhibitor RNAsin (40 U/μL) (Promega, Madison, WI, USA), 0.8 μL of M-MLV RT 200 U/μL and 5 ΜL of RNA template for a final reaction volume of 20 μL. The reaction mixture was then incubated at 25 °C for 15 min, followed by 37 °C for 1 h and final denaturation at 94 °C for 5 min. The cDNA was stored at –20 °C until use [19].
Quantitative real-time RT-PCR (rRT-qPCR)
The thermostability of each viral RNA was assessed by rRT-qPCR assays based on SYBR Green I. The rRT-qPCR assays were performed on the LightCycler 2.0® instrument (Roche Applied Science, Mannheim, Germany).
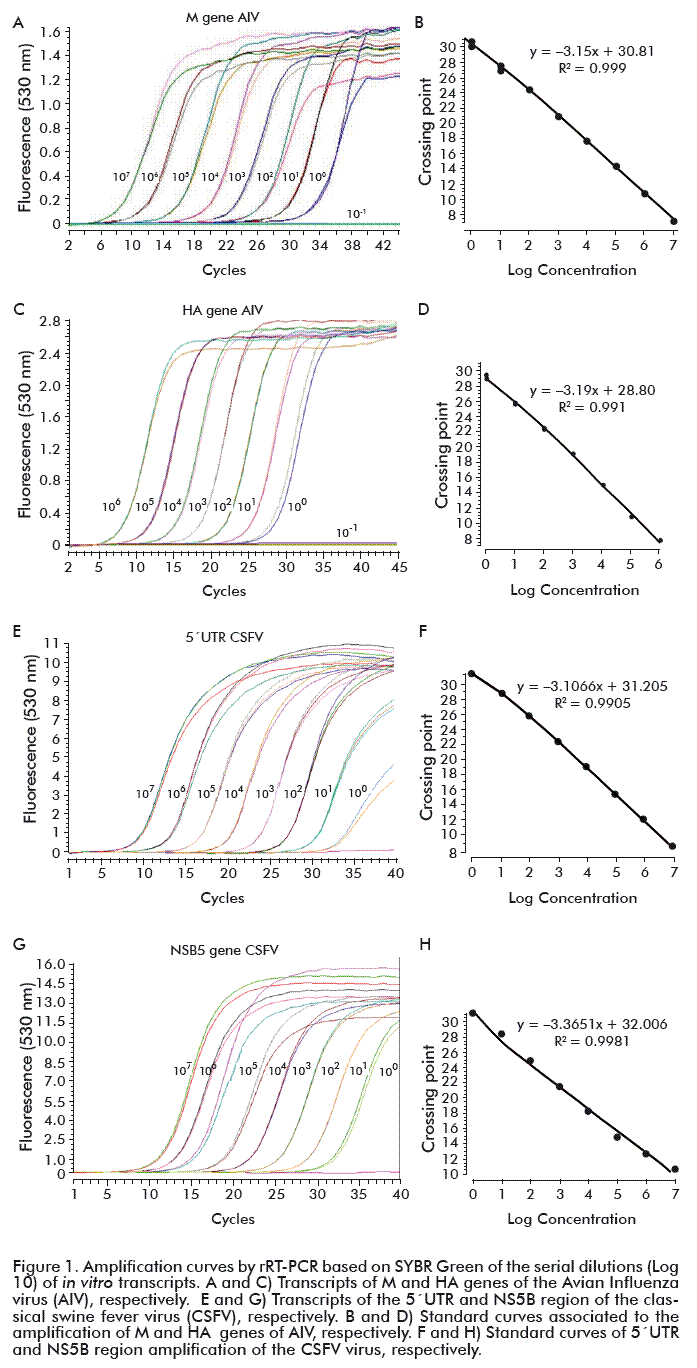
To quantify RNA concentrations in terms of copy number, from the different target regions under study, standard curves were generated. Each standard curve was obtained by testing sequential ten-fold dilutions of the in vitro-transcribed RNA, from the 5´UTR region and the NS5B protein of the CSFV RNA and the M and HA genes of AIV RNA, in nuclease free water (Promega, Madison, USA).
The ‘in vitro’ transcripts in each case were obtained using the commercial kit MEGAscript_Kit (Ambion), according to the manufacturer’s recommendations. The efficiency of each reaction as well as the linearity, amplification efficiency and dynamic range of each generated curve were calculated using the Light Cycler 2.0 software (Version 4.05).
The target regions for the specific detection of the viral RNA of the CSFV were the 5´UTR and that of the NS5B protein. The rRT-qPCR corresponding to the 5´UTR region was carried out with the working protocol proposed by Hoffmann et al. [20], with the primer pair CSF100-F: 5´ATGCCCAYAGTAGGACTAGCA3´ and CSF192-R: 5´CTACTGACGACTGTCCTGTAC3´) and probe FAM-TGGCGAGCTCCCTGGGTGGTCTAAGT-TAMRA). Meanwhile, the rRT-qPCR of the NS5B region was performed by the protocol described by Perez et al. [21], with the primer pair (CSFV1: 5´CCTGAGGACCAAACACATGTTG3´ and CSFV2: 5´TGGTG-GAAGTTGGTTGTGTCTG3´) proposed by Díaz de Arce et al. [22].
Target regions for the specific detection of AIV type A viral RNA were a fragment of the matrix gene (M gene) and a fragment of the H5 subtype of the hemagglutinin gene (HA gene). The primers used for the specific detection of the M gene and the HA gene were those previously published by Spackman et al. [23] (M+25- 5´-TGAGTCTTCTAACCGAGGTCG-3´; M-124-5´- TGCAAAGACACTTTCCAGTCTCTG-3´) and by Lee et al. [24] (H5155f: 5´-ACACATGCYCARGACATACT-3´; H5699r: 5´-CTYTGRTTYAGTGTTGATGT-3´) respectively.
In the rRT-qPCR assay of the M gene the reaction was run in a final volume of 20 μL, containing a concentration of 0.4 μM of the sense and the antisense primers, 2 mM MgCl2, 2 μL of FastStart DNA Master SYBR Green I (10×), 5 μL of cDNA template and nuclease-free water. The reaction was run under the following conditions: 10 min at 95 °C, followed by 40 cycles at 95 °C for 10 s, 55 °C for 10 s and 72 °C for 20 s. After the PCR cycles, a specific melting curve was generated (0 sec at 95 °C, 15 sec. at 65 ° C, a ramp time of 20 °C/s. and 0 s at 95 °C with a ramp time of 0.1 °C/s) in order to discriminate between products of specific and non-specific amplifications. Quantification of the HA gene of the H5 subtype AIV was performed by the rRT-PCR assay described by Pérez et al. [25].
Statistical analysis
Data were analyzed using the statistical package InfoStat 2016 [26], where a simple variance and mean contrast analysis was performed by Duncan’s multiple-range test.
RESULTS AND DISCUSSION
RNAs are highly labile molecules and rapidly degrade under inappropriate storage conditions [2]. A large variety of DNA/RNA biological samples and positive controls of molecular assays preserved at ultra-low temperatures (–20 or –80 ºC), are lost each year in many laboratories due to temporary absence of power, either as a result of damages in energy-generating systems or natural disasters. As example there could be mentioned the million dollar losses in bio-specimens caused by Hurricane Sandy in US in 2012 [27, 28].
Both dehydration and cryopreservation keep the nucleic acid in a crystallized state where the molecule has very few levels of motion and the likelihood of chemical reactions is practically unlikely [29]. It has been shown that even nucleic acids preserved at ultra-low temperatures have some reactivity when they are in a hydrated environment. This was demonstrated by Ma et al. [30], where they observed that ribonucleases continue to be active in frozen RNA at –20 ºC. Also, other authors suggest that the activity of some ribozymes remains significant at –70 ºC [31]. In more recent studies on the conservation of dehydrated RNA, it has been shown that with prior treatment with stabilizing solutions, they can be stable even if they are preserved at room temperature [7, 32, 33].
To demonstrate that a method for preserving nucleic acid stability is effective, the conservation of the strand structure must be verified over time. The most commonly used method for this verification was the RT-qPCR [5, 34], where the amplification efficiency of the RT-PCR assays must be taken into account through a standard curve [35, 36]. The standard curve for both viral agents was established from in vitro transcripts, since they are an alternative for the development and evaluation of molecular tools, by guaranteeing a positive control of intact RNA, a critical aspect for screening [7, 25].
The standard curves obtained for the quantification of AIV M and HA genes and the 5’UTR and NS5B regions of the CSFV (Figure 1) showed amplification efficiency between 1.8 and 2.0, these values permissible for the real time PCR assays with the use of Roche’s Light Cycler [37]. The coefficient of determination in all cases was R2 > 0.99. Taking into account that the determination coefficient expresses the measure of the variation of the data, which is explained by the linear relationship between the two variables (initial copy number of the genes and the threshold cycle (Ct)) one can expect good certainty in the quantification of the samples to evaluate.
The standard curve in terms of RNA copy number obtained for the quantification of M transcript showed a linear range from 107 to 100 copies/μL and from 106 to 100 copies/μL for the HA transcript (Figure 1A and C). On the other hand, for the target regions evaluated for CSFV, the linear range obtained for each 5´UTR and NS5B transcript generated standard curves from 107 to 100 copies/μL (in terms of RNA copy number) in both cases (Figure 1 E and G).
In the evaluation of the CSFV RNA thermostability, the target regions of the viral RNA that are commonly used in the diagnosis of this viral entity were selected. For the diagnosis of CSFV, some laboratories target the 5´UTR region [16, 38, 39], as it has a highly conserved sequence among the Pestiviruses since the internal ribosome entry site (IRES) is located in this region, which is indispensable for the initiation of the translation of viral proteins [40]. However, other laboratories target the NS5B protein binding region [19, 41], which is a region located at the C-terminal of the polyprotein that binds to the 3´UTR end of the viral RNA. This protein plays a relevant role in the replication of the viral genome. It is known that problems in this region of the genome would cause degradation of all viral RNA, with subsequent reduction of viral replication [42].
The results obtained in the evaluation of the thermostability of the preserved CSFV RNA in dry pellet form (Figure 2) showed that both target regions 5´UTR and NS5B had a very similar behavior throughout the experiment. Although there were statistically significant differences between the thermostability of each target region depending on the storage temperature after 30 days in none of the cases the DNA amplification performance of the samples was drastically affected. For both targets up to the end of the experiment, quantifiable values above 102 copies/μL of RNA were detected, even in those samples that were stored at the most extreme temperature (37 ºC). These results show that laboratories that store or receive the CSFV RNA in the form of dry pellet can use both targets for the diagnosis of this disease, since under these conservation conditions both fragments of the genome are thermostable for at least 60 days.
Several rRT-PCR assays have been described and validated for the diagnosis of AIV to amplify both, regions of the matrix gene (M) and the HA2 region of the HA gene. The M gene is preserved in all 16 subtypes of the virus from all geographic regions, making it ideal for AIV detection. While the HA2 region is relatively preserved among the hemagglutinin genes, which makes it the most relevant for the diagnosis of different viral subtypes [23, 43-45]. Highly pathogenic avian influenza viruses (HPAI) have been associated with H5 and H7 subtypes, although not all viruses of these subtypes cause HPAI. Both, HPAI and low pathogenic avian influenza (LPAI) cause a highly contagious disease capable of spreading to susceptible populations in a short period of time [46]. This can have devastating effects on the poultry industry, particularly if it occurs in high bird density areas [47]. The most effective strategy to fight efficiently the AIV is to conduct early detection and warning to prevent the spread of the disease and achieve effective control. Hence, the preservation of AIV RNA stability in these two diagnostic regions (M and HA gene) is indispensable for the accurate diagnosis of this entity.
It is important to note that the AIV genome consists of eight segments (PB2, PB1, PA, HA, NP, NA, M and NS) coding for nine proteins (PB2, PA, HA, NP, NA, M1 and M2, NS1 and NS2) [48, 49]. The degradation of one of these segments of the genome can be translated into possible failures in the viral multiplication cycle and thus subvert viral isolation [49]. Nevertheless, if degradation does not occur at the recognition sites of the primers in the segment used for the diagnosis, hence, the performance of the molecular assays is not affected.
There were also found differences in the behavior of the thermostability of the AIV M and HA genes (Figure 3). Even though both target regions of the genome generated a quantifiable amplification of the RNA by the rRT-qPCR throughout the experiment, the yield of amplification of the target region of the HA gene was gradually decreasing over time. Notably, that yield was further affected when the RNA was stored as dry pellet at 37 °C. This same behavior was not observed in the yield of the M gene amplification, which was very similar at all time points evaluated, regardless of the samples storage temperature.
As we have observed, the conservation of RNA as dry pellets within hermetically sealed polyethylene tubes could be an efficient and low cost alternative for the preservation of viral RNAs that are stored for long periods in diagnostic laboratories and biobanks. It has been shown that nucleic acids stored in a dehydrated matrix at ultra-low temperatures can be preserved for hundreds of years [31]. In most laboratories where genetic material is preserved, there are storage conditions of –20 or –80 °C, and unexpected electrical power failures can occur. In that setting, the preservation method proposed in this work guarantee that viral RNAs stored at ultra-low temperatures can continue its storage once they reach room temperature, without degradation in the form of dry pellet and for up to two months. Thus, losses of stored genetic material would be avoided. Above all, the loss of positive controls would not be regretted. These controls are indispensable for the execution of molecular tests and commonly difficult to obtain, especially in those diseases that are exotic to a country.
In summary, the study demonstrated that storing viral RNAs under dry pellet conditions within hermetically sealed polyethylene tubes is an effective alternative for the preservation of viral RNA stability for long periods of time at room temperature.
REFERENCES
1. Ivanova NV, Kuzmina ML. Protocols for dry DNA storage and shipment at room temperature. Mol Ecol Resour. 2013;13(5):890-8.
2. Seelenfreund E, Robinson WA, Amato CM, Tan AC, Kim J, Robinson SE. Long term storage of dry versus frozen RNA for next generation molecular studies. PLoS One. 2014;9(11):e111827.
3. Gonzalez-Perez I, Rosa IG, Cayarga AA, Hernandez YP, Gonzalez YJ, Victores YR, et al. Scaling up in vitro transcription synthesis of RNA standards for competitive quantitative RT-PCR: looking for bigger yields. Anal Biochem. 2009;385(1):179-81.
4. Oivanen M, Kuusela S, Lonnberg H. Kinetics and mechanisms for the cleavage and isomerization of the phosphodiester bonds of RNA by Bronsted acids and bases. Chem Rev. 1998;98(3):961-90.
5. Fabre AL, Colotte M, Luis A, Tuffet S, Bonnet J. An efficient method for long-term room temperature storage of RNA. Eur J Hum Genet. 2014;22(3):379-85.
6. Shinriki N, Ishizaki K, Miura K, Ueda T, Harada F. Degradation of nucleic acids with ozone. III. Mode of ozone-degradation of mouse proline transfer ribonucleic acid (tRNA) and isoleucine tRNA. Chem Pharm Bull (Tokyo). 1983;31(10):3601-8.
7. Hoffmann B, Depner K, Schirrmeier H, Beer M. A universal heterologous internal control system for duplex real-time RT-PCR assays used in a detection system for pestiviruses. J Virol Methods. 2006;136(1-2):200-9.
8. Martinez H, Beaudry G, Veer J, Robitaille M, Wong D, Iverson B, et al. Ambient temperature storage of RNA in GenTegra™ for use in RT-qPCR. BioTechniques. 2010;48(4):328-9.
9. Moscoso H, Raybon EO, Thayer SG, Hofacre CL. Molecular detection and serotyping of infectious bronchitis virus from FTA filter paper. Avian Dis. 2005;49(1):24-9.
10. Spackman E, Pedersen JC, McKinley ET, Gelb J. Optimal specimen collection and transport methods for the detection of avian influenza virus and Newcastle disease virus. BMC Vet Res. 2013;9:35.
11. Floegel-Niesmann G, Bunzenthal C, Fischer S, Moennig V. Virulence of recent and former classical swine fever virus isolates evaluated by their clinical and pathological signs. J Vet Med B Infect Dis Vet Public Health. 2003;50(5):214-20.
12. World Organization of Animal Health. Avian influenza. In: OIE. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. Paris: OIE; 2014. p. 1128-38.
13. Depner K, Hoffmann B, Beer M. Evaluation of real-time RT-PCR assay for the routine intra vitam diagnosis of classical swine fever. Vet Microbiol. 2007;121(3-4):338-43.
14. Cheng D, Zhao JJ, Li N, Sun Y, Zhou YJ, Zhu Y, et al. Simultaneous detection of Classical swine fever virus and North American genotype Porcine reproductive and respiratory syndrome virus using a duplex real-time RT-PCR. J Virol Methods. 2008;151(2):194-9.
15. Zambon M, Goddard N, Meijer A, McCauley J, Daniels R. Diagnostic preparedness in Europe for detection of avian influenza A(H7N9) viruses, Techni-cal briefing note. Stockholm: European Centre for Disease Prevention and Control, ECDC; 2013.
16. Haines FJ, Hofmann MA, King DP, Drew TW, Crooke HR. Development and validation of a multiplex, real-time RT PCR assay for the simultaneous detection of classical and African swine fever viruses. PLoS One. 2013;8(7):e71019.
17. Reid SM, Ferris NP, Hutchings GH, Zhang Z, Belsham GJ, Alexandersen S. Detection of all seven serotypes of foot-and-mouth disease virus by real-time, fluorogenic reverse transcription polymerase chain reaction assay. J Virol Methods. 2002;105(1):67-80.
18. Fernandez-Pinero J, Gallardo C, Elizalde M, Robles A, Gomez C, Bishop R, et al. Molecular diagnosis of African Swine Fever by a new real-time PCR using universal probe library. Transbound Emerg Dis. 2013;60(1):48-58.
19. Diaz de Arce H, Perez LJ, Frias MT, Rosell R, Tarradas J, Nunez JI, et al. A multiplex RT-PCR assay for the rapid and differential diagnosis of classical swine fever and other pestivirus infections. Vet Microbiol. 2009;139(3-4):245-52.
20. Hoffmann B, Beer M, Schelp C, Schirrmeier H, Depner K. Validation of a real-time RT-PCR assay for sensitive and specific detection of classical swine fever. J Virol Methods. 2005;130(1-2):36-44.
21. Perez LJ, Diaz de Arce H, Tarradas J, Rosell R, Perera CL, Munoz M, et al. Development and validation of a novel SYBR Green real-time RT-PCR assay for the detection of classical swine fever virus evaluated on different real-time PCR platforms. J Virol Methods. 2011;174(1-2):53-9.
22. Diaz de Arce H, Nunez JI, Ganges L, Barreras M, Frias MT, Sobrino F. An RT-PCR assay for the specific detection of classical swine fever virus in clinical samples. Vet Res. 1998;29(5):431-40.
23. Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, et al. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol. 2002;40(9):3256-60.
24. Lee MS, Chang PC, Shien JH, Cheng MC, Shieh HK. Identification and subtyping of avian influenza viruses by reverse transcription-PCR. J Virol Methods. 2001;97(1-2):13-22.
25. Perez LJ, Diaz de Arce H, Cilloni F, Salviato A, Marciano S, Perera CL, et al. An SYBR Green-based real-time RT-PCR assay for the detection of H5 hemagglutinin subtype avian influenza virus. Mol Cell Probes. 2012;26(3):137-45.
26. Di Rienzo JA, Casanoves F, Balzarini MG, Gonzalez L, Tablada M, Robledo CW. InfoStat version 2016. National University of Cordova, Argentina: Grupo InfoStat, FCA. 2016 [cited 2016 Jan 17]. Available from: http://www.infostat.com.ar
27. CBS. News Staff. Freezer Malfunction Thaws 150 Brains at Harvard Research Hospital. CBS NEWS. 2012 Jun 1 [cited 2016 Jan 17]. Available from: http://www.cbsnews.com/news/freezer-malfunction-thaws-150-brains-at-harvard-research-hospital
28. Kuntzman G. Loses years of scientific research and thousands of mice to hurricane Sandy. NY Daily News. 2012 Oct 30 [cited 2016 Jan 17]. Available from: http://www.nydailynews.com/new-york/nyu-loses-thousands-mice-sandy-article-1
29. Lee SB, Crouse CA, Kline MC. Optimizing Storage and Handling of DNA Extracts. Forensic Sci Rev. 2010;22(2):131-44.
30. Ma S, Huang Y, van Huystee RB. Improved plant RNA stability in storage. Anal Biochem. 2004;326(1):122-4.
31. Seyhan AA, Burke JM. Mg2+-independent hairpin ribozyme catalysis in hydrated RNA films. RNA. 2000;6(2):189-98.
32. Stevens DS, Crudder CH, Domingo GJ. Post-extraction stabilization of HIV viral RNA for quantitative molecular tests. J Virol Methods. 2012;182(1-2):104-10.
33. Conny M, Wusheng Y, Rodrigo C, Amy PN, Skubitz A, et al. Short-Term Stability Study of RNA at Room Temperature. Biopreserv Biobank. 2012;10(6):532-42.
34. Liu X, Li Q, Wang X, Zhou X, He X, Liao Q, et al. Evaluation of DNA/RNAshells for room temperature nucleic acids storage. Biopreserv Biobank. 2015;13(1):49-55.
35. Aguilera P, Ruiz M, Rocha M, Pineda B, Chánez ME. PCR en tiempo real. In: Cornejo A, Serrato A, Rendón B, Roche MG, editors. Herramientas moleculares aplicadas en ecología: aspectos teóricos y prácticos. Ciudad de México: Laboratorio de Patología Vascular Cerebral, Instituto Nacional de Neurología y Neurocirugía; 2014 [cited 2016 Jun 16]. Available from: http://www2.inecc.gob.mx/publicaciones/libros/710/pcrtiempo.pdf
36. Wong ML, Medrano JF. Real-time PCR for mRNA quantitation. Biotechniques. 2005;39(1):75-85.
37. Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30(9):e36.
38. Liu J, Fan XZ, Wang Q, Xu L, Zhao QZ, Huang W, et al. Dynamic distribution and tissue tropism of classical swine fever virus in experimentally infected pigs. Virol J. 2011;8:201.
39. Hsu WL, Chen CL, Huang SW, Wu CC, Chen IH, Nadar M, et al. The untranslated regions of classic swine fever virus RNA trigger apoptosis. PLoS One. 2014;9(2):e88863.
40. Xiao M, Zhu ZZ, Liu J, Zhang CY. Prediction of recognition sites for genomic replication of classical swine fever virus with information analysis. Mol Biol. 2002;36(1):48-57.
41. Malswamkima D, Rajkhowa TK, Chandra R, Dutta TK. Pathology and molecular diagnosis of classical swine fever in Mizoram. Vet World. 2015;8(1):76-81.
42. Haegeman A, Dewulf J, Vrancken R, Tignon M, Ribbens S, Koenen F. Characterisation of the discrepancy between PCR and virus isolation in relation to classical swine fever virus detection. J Virol Methods. 2006;136(1-2):44-50.
43. United States Department of Agriculture. Avian influenza testing and diagnostics. Washington, D.C.: USDA Press Office; 2015.
44. Lee CW, Suarez DL. Application of real-time RT-PCR for the quantitation and competitive replication study of H5 and H7 subtype avian influenza virus. J Virol Methods. 2004;119(2):151-8.
45. Perera CL, Díaz de Arce H, Pérez LJ. Actualización y perspectivas en el diagnóstico del virus de la influenza aviar. Rev Salud Anim. 2011;33(1):1-7.
46. Heine HG, Foord AJ, Wang J, Valdeter S, Walker S, Morrissy C, et al. Detection of highly pathogenic zoonotic influenza virus H5N6 by reverse-transcriptase quantitative polymerase chain reaction. Virol J. 2015;12:18.
47. McAuley JL, Chipuk JE, Boyd KL, Van De Velde N, Green DR, McCullers JA. PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 2010;6(7):e1001014.
48. Lekcharoensuk P, Nanakorn J, Wajjwalku W, Webby R, Chumsing W. First whole genome characterization of swine influenza virus subtype H3N2 in Thailand. Vet Microbiol. 2010;145(3-4):230-44.
49. Palase P, Shaw ML. Orthomyxoviridae: The viruses and their replication. In: Knipes D, Howley P, editors. Field’s Virology. 5th edition. Philadelphia: Lipincott Williamson and Wilkinson; 2007. p. 1647-89.
Received in July, 2016.
Accepted in November, 2016.
Damarys Relova. Departamento de Virología, Centro Nacional de Sanidad Agropecuaria, CENSA. Apartado 10, San José de las Lajas, Mayabeque, Cuba. E-mail: drelova@censa.edu.cu.


{kind=link}
{kind=link}
{kind=link}