










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.34 no.4 La Habana Oct.-Dec. 2017
RESEARCH
Genetic stability of the Primary Cell Bank expressing the recombinant antigen MY32/Ls of Sea lice
Estabilidad genética del Banco de Células Primario que expresa el antígeno recombinante MY32/Ls del piojo de mar
Niuvis Montoya1, Nemecio González1, Ileana Sánchez1, Rafael Pimentel1, Liliana Basabe2, Rosa M Basulto1, Liszoe Galdós1, Nestor Mora1, Eladio Salazar1, Diasmarys Salinas1, Carlos Pérez1, Alain Moreira1, Rutdali Segura1
1 Centro de Ingeniería Genética y Biotecnología de Camagüey. Circunvalación Norte y Ave. Finlay, Camagüey, Camagüey, Cuba.
2 Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 entre 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.
ABSTRACT
The recombinant MY32/Ls protein is used as a vaccine antigen against sea lice Lepeophtherius salmonis. It is produced from the fermentation of the E. coli BL21 (DE3)-pET28a-my32/Ls expression system, which is stored frozen at -70 ºC in a pure and homogeneous Primary Cell Bank (PCB). This work was aimed to analyze the genetic stability of cells from this PCB grown for a high number of generations, and how it influences cell growth rate, doubling time and the expression of the protein of interest, either in shake flasks or cultured in a 200-L bioreactor. It was found that in this large scale process, the strain replicates for 46 generations. It was demonstrated that the number of generations did not affect the growth rate (μ) and the doubling time. Similarly, plasmid stability and protein expression remained unaffected and a 100 % identity match was obtained for the my32/Ls gene sequences in the expression plasmid isolated from PCB samples and after nearly 100 generations. It was also shown that the expression system of the MY32/Ls protein is genetically stable throughout the production process, supporting the development of a working cell bank from the characterized PCB that guarantees the stability, integrity and safety of large scale productions of this recombinant protein.
Keywords: plasmid stability, primary cell bank, recombinant protein production, number of generations, sea lice, MYL32/Ls.
RESUMEN
La proteína MY32/Ls recombinante se emplea como antígeno vacunal contra el piojo de mar Lepeophtherius salmonis. La misma se produce a partir de la fermentación del sistema de expresión en la cepa bacteriana de Escherichia coli BL21(DE3)-pET28a-my32/Ls, la cual se preserva congelada a –70 ºC en un Banco de Células Primario (BCP) puro y homogéneo. El objetivo del presente trabajo fue determinar la estabilidad genética del BCP mediante en células crecidas propagadas durante un alto número de generaciones, así como evaluar su influencia sobre el crecimiento y la expresión de la proteína. Al cultivo con elevado número de generaciones se le analizó la estabilidad plasmídica, la expresión de la proteína recombinante, y el patrón de restricción enzimática y se secuenció el gen de interés. Se determinó que las la cepa se propaga durante 46 generaciones durante el proceso productivo en un fermentador de 200-L. Se comprobó que el número de generaciones no afectó la velocidad de crecimiento (µ) ni el tiempo de duplicación durante todo el estudio. La estabilidad plasmídica y la expresión de la proteína recombinante se mantuvieron a niveles equivalentes entre el BCP y la cepa propagada durante casi 100 generaciones, y se obtuvo una coincidencia del 100 % de la identidad plasmídica del gen my32/Ls entre ambos cultivos. El patrón de restricción obtenido coincidió con el esperado para el plásmido recombinante. El estudio permitió conocer que el sistema de expresión de la proteína MY32 es genéticamente estable durante todo el proceso productivo, lo que posibilita la elaboración de un Banco de Células de Trabajo que garantice la estabilidad, la integridad y la seguridad de las producciones a gran escala de la proteína MY32/Ls.
Palabras clave: estabilidad plasmídica, banco de células primario, producción de proteína recombinante, número de generaciones, piojo de mar, MYL32/Ls.
INTRODUCTION
Sea lice Caligus rogercresseyi and Lepeophtherius salmonis have emerged as the main pathogen affecting the salmon industry during the last 30 years [1, 2]. This has led to the introduction of a wide range of treatments to try to contain or control the infection. However, pathogens’ resistance to chemicals, and the associated environmental damage and high costs associated to those compounds redirected the attention to more enduring treatments, such as vaccination [3]. Vaccination strategies under research against sea lice follows other approaches common to veterinary vaccines against insect-mediated infestations, like those employing tick gut antigens (Bm86 protein) for vaccination in cattle and marketed as Gavac® and TickGARD® [3], aimed to reduce larval populations [4]. Specifically for sea lice, some groups have reported the potential of the MY32 protein as vaccine antigen, this protein playing a physiological role in reproduction and able to induce a protecting response in salmon upon vaccination (57 % inhibition of C. rogercresseyi and L. salmonis infestation).
In this regard, our group have recently established an expression system in Escherichia coli BL21(DE3)-pET28a-my32/Ls, expressing the MY32/Ls intracellularly and forming inclusion bodies [3, 5]. The production process of this protein for vaccine purposes includes the storage of the expression system in a Primary Cell Bank (PCB) by freezing at –70 ºC. This step is fundamental for the construction and characterization of recombinant products, particularly to guarantee their high stability, integrity and safety for biopharmaceutical application after the large scale production process [6].
In this regard, the analysis of genetic stability of the cell bank determines the integrity of the genetic sequence coding for the recombinant protein and its stability until the end of the production process [7]. Stability could be affected after cloning due to sequence modification in cells upon culture after several generations [8, 9], mainly in large scale cultures [10]. It is essential to study the stability of the expression vector in the PCB during the scale-up of the protein production process, and for several generations. Therefore, it was evaluated the genetic stability of the PCB expressing the recombinant protein MY32/Ls, which determines the consistency of the process and the final product. It was analyzed through the study of the cell population doubling of the Escherichia coli BL21(DE3)-pET28a-my32/Ls expression system during the production process, which was modeled at a 3-L scale and equivalent to that of a 200-L production fermentation. This establishes whether the expression system retains its expected stability and if it expresses the recombinant antigen properly.
MATERIALS AND METHODS
Bacterial strain and expression vector
The E. coli BL21(DE3) (genotype F–, ompT, hsdS (r B–, mB–) gal, Δ lon, dcm (λD69) strain [11] was used transformed with the plasmid pET28a-my32/Ls [3]. This constructs expresses the T7 RNA polymerase under the control of the lacUV5 promoter, and the gene of interest cloned under the control of the T7 promoter, with a plasmid size of 5369 bp. The protein of interest is then induced by adding IPTG to the culture medium. The vector also carries a kanamycin resistance gene and a 6His tag for downstream protein purification. The gene MY32/Ls codes for the Akirin 2 protein of Lepeophtheirus salmonis, which is expressed intracellularly. It was previously isolated by reverse transcription and PCR as cDNA.
Culture media and solutions
Luria-Bertani (LB) medium was prepared for propagation (5 g/L Yeast extract (Oxoid, Germany); 10 g/L Tryptone (Oxoid, Germany), 5 g/L NaCl (Merck, Germany); pH 7.3). Solid LB media was also prepared by adding Technical Agar #3 1.5 % w/v. Both media were autoclaved at 121 ºC for 30 min.
Solutions of 40 % Glucose (Sigma, USA) and 50 mg/mL Kanamycin (Applichem, Germany) were made, and subsequently sterilized at 121 ºC for 20 min or filtered, respectively. They were added to the LB liquid culture medium upon use and when reaching 45-50 ºC to the solid culture medium (12.5 mL Glucose 40 % and 400 μL of 50 mg/mL Kanamycin per liter of medium).
Protein expression was analyzed by using the fermentation medium described by Studier [11].
Estimation of bacterial number of generations at 200-L scale and design of the genetic stability study at laboratory scale
The number of generations of the transformed bacterial strain E. coli BL21 (DE3)-pET28a-my32/Ls was studied at 200-L scale representing the proposed working volume for the production of the recombinant MY32/Ls protein. For this, the wet weight and cellular viability parameters were proportionally estimated at 3-L scale. The number of generations was calculated as described for a culture at exponential growth phase [12].
Bacterial culture at 3-L scale with IPTG-induced protein expression
The E. coli BL21 (DE3)-pET28a-my32/Ls strain was cultured in a 3-L Marubishi bioreactor (Marubishi, Japan) at the following conditions: 1 vvm aeriation, 500 rpm stirring, 37 ± 1 ºC, and culture medium pH 7.0-7.2.
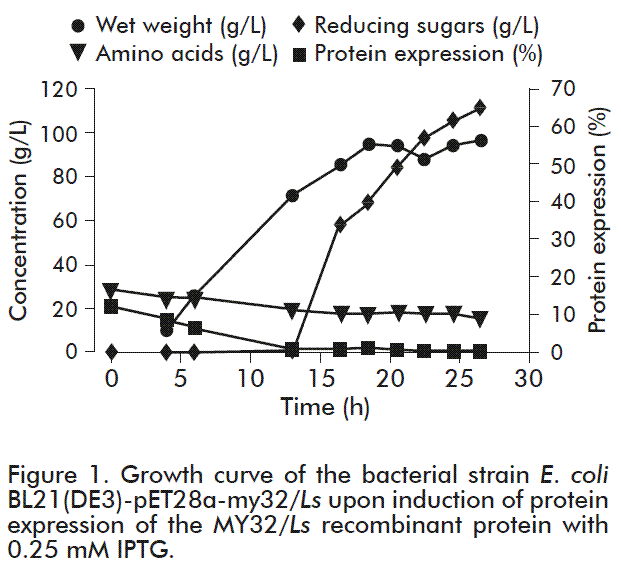
Fermentation was done in two phases, the first comprising a batch culture for 7 h, followed by the second consisting on growth by fed-batch fermentation on pH demand. For this second phase, the feed medium was supplied through the pH pump (acid pH) to reach a high density cell culture with pH values within the established range. Samples were periodically taken to evaluate cell growth, the expression of the protein of interest, and to monitor glucose and amino acids concentration. Once reaching the desired wet weight values, the expression of the MY32/Ls protein was in-duced with 0.25 mM IPTG for 12 h [13].
Estimation of the number of generations for the E. coli BL21 (DE3)-pET28a-my32/Ls strain at 200-L scale in 500-mL flasks
The number of generations of the E. coli BL21 (DE3)- pET28a-my32/Ls strain at the 200-L fermentation scale was estimated considering the following parameters:
1. The wet weight and cell viability values reached after the induction of protein expression with IPTG at 3-L, which were used to estimate the biomass (g) and total cell number, respectively, that could be obtained in a 200-L bioreactor.
2. The number of viable cells calculated per g of biomass.
Then, the number of cell generations that could be reached during the production process was calculated, and a shake flasks culture process was designed, at the end of which the value for this this kinetic parameter will be doubled.
Design of the assay to determine the number of cell generations for the E. coli BL21 (DE3)- pET28a-my32/Ls at laboratory scale
An assay was designed in shake flasks at laboratory scale to determine the number of cell generations for the E. coli BL21 (DE3)-pET28a-my32/Ls. Three cell propagation cycles (C1 to C3) were established, consisting on inoculation from Petri dishes to 500-mL Erlenmeyer flasks and then to Petri dishes for c.f.u. count.
E. coli BL21 (DE3) cells transformed with the plasmid pET28a-my32/Ls were inoculated from a cell storage vial, containing Luria-Bertani (LB) storage medium with 20 % glycerol, onto three dishes containing solid LB medium, and they were further incubated for 24 h at 37 ºC. Then, three Erlenmeyer flasks containing 500 mL of LB medium each were inoculated from five colonies each and they were incubated at 37 ºC for 8 h. After that, wet weight and viability were determined to each culture.
Evaluation of the genetic stability of the pET28a-my32/Ls construct in E. coli BL21(DE3) fermentations duplicating the number of generations of the production process
Influence of the number of cell generations on bacterial strain growth
The influence of cell generation number on strain growth was studied through the specific growth rate (μ) and doubling time (dT) at 37 ºC for 250 rpm. Samples were taken for the first and third propagation cycles (C1 and C3, respectively) at 0, 2, 4, 5, 6, 7 and 8 h and optical density was measured at 600 nm (O.D.600 nm). Both parameters were determined as previously described [14].
Plasmid stability testing
Plasmid stability was checked at the end of the third fermentation cycle as described [15] at the end of the third propagation cycle (C3). Colonies were sampled in plates containing unsupplemented solid LB medium, and also supplemented with kanamycin. Plates were incubated at 37 ºC for 24 h.
Determination of MY32/Ls protein expression in shake flasks
Protein expression was determined in bacterial cultures corresponding to the bacterial strain with high generation number by SDS-PAGE in 12 % gels under reducing conditions [16].
Purification of the pET28a-my32/Ls plasmid DNA
Ten bacterial colonies with 100 cell generations (C3 cycle) were randomly selected, and their plasmid DNA molecules were purified with the Wizard® Plus Midipreps DNA Purification System (Promega, USA). The resulting plasmid material was checked by restriction with Nco I-Hind III, Hind III and Nco I. Restriction patterns were checked by 0.8 % agarose gel electrophoresis.
my32/Ls gene sequencing
The fragment in the construct pET28a-my32/Ls containing the gene my32/Ls was sequenced by using the DNA primers T7p (5´-TAATACGACTCACTATAGGG-3´) and T7t (5´-GCTAGTTATTGCTCAGCGG-3´), which were specific for the pT7 promoter and T7 terminator sequences, respectively. The fragment was sequenced in triplicate, by the Big Dye Chemistry system with capillary electrophoresis (Applied Biosystems; Macrogen) and processed in a ABI 3730XL Genetic Analyzer.
Genetic sequence data of the samples from the high generation number were processes with the Vector NTI Suite software (version 8.0); the consensus sequence was generated from sequencing replicates, and it was further aligned to the expected standard plasmid sequence sequence established for the PCB of the E. coli BL21 (DE3)-pET28a-my32/Ls.
Statistical analysis
Data obtained were analyzed with the aid of the STATGRAPHICS Plus 5.1 statistical package. Paired samples comparisons and the typical deviation measures were assessed by the Student’s t tests and Fisher’s exact test, respectively.
RESULTS
Estimation of generation number for fermentation at 200-L and plasmid genetic stability at laboratory scale
E. coli BL21(DE3)-pET28a-my32/Ls culture at 3-L induced with IPTG
Culture reached a 70 g/L cell density after 13 h (Figure 1), and then, expression of the MY32/Ls protein was induced with 0.25 mM IPTG and carried out for 12 h until reaching 90 g/L of wet biomass at the end of culture. Protein expression was achieved for up to 63.29 %, as determined by densitometry analysis of PAGE gels.
Assessment of generation number of the E. coli BL21(DE3)-pET28a-my32/Ls strain in shake flask cultures and during the production process at 200-L culture
The number of cell generations able to be achieved by the E. coli BL21(DE3)-pET28a-my32/Ls strain in a 200-L bioreactor was calculated according to viability and wet weight, which were determined during the starting shake flasks propagation (C1) (Table). It was estimated to reach a cell mass of 100 g/L and a cell density of 1.1 × 1015 total cells in the bioreactor under the previously described conditions, and up to 45.6 generations. With three successive propagation cycles C1 to C3 (Petri dishes-flasks-Petri dishes) nearly 100 generations could be achieved.
Evaluation of the genetic stability of the plasmid construct in fermentations duplicating the generation number of the production process
Influence of generation number on bacterial strain growth
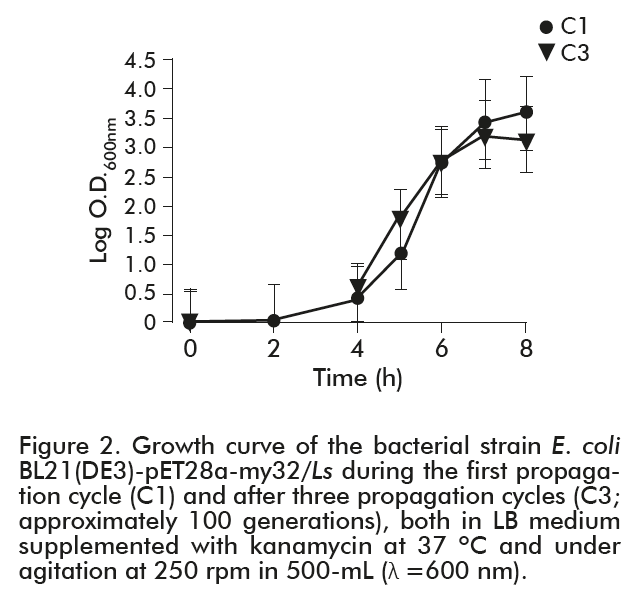
As shown in figure 2, the bacterial strain reached the exponential phase during the last propagation after 5 h of culture (1.21 O.D.600 nm), while the same phase was reached during the first propagation after 4 h (0.558 O.D.600 nm). Noteworthy, exponential phase ended in both cultures after 6 h, with 2.0 O.D.600 nm. This evidenced that the number of generations reached by the bacterial strain did not affected the metabolic processes needed for the cellular division.
The specific growth rate of the strain during the first propagation cycle was 0.78 h-1, with a doubling time of 53 min, while the same parameters were 0.81 h-1 and 51 min, respectively, for the C3 cycle, with no statistically significant differences between the specific growth rate mean values. These results demonstrated that the number of generations did vary neither the specific growth rate, nor the doubling time of the strain.
These two parameters were only assessed between propagation cycles 1 (34 cell generations) and 3 (100 cell generations), to analyze the possible influence of generation number on growth kinetics parameters.
Plasmid stability
The pET28a-my32/Ls plasmid was shown to be 98 % stable within the E. coli BL21(DE3) strain after approximately 100 cell generations (Figure 3).
Assessment of MY32/Ls protein expression in shake flasks
As shown in figure 4, the cell generation number did not affected the MY32/Ls protein expression, with a notable protein band detected in cell extracts of shake flask cultures in the 20-28 kDa molecular weight range by SDS-PAGE (Figure 4A), coincident with the migration pattern of the standard MY32/Ls protein on lane 1. This pattern was similar to that observed for the bands shown in figure 5B [3] after 100 cell generations. The MY32/Ls protein band was absent in the cell extracts from non-induced cell cultures (data not shown). Up to 56.92 % of expression was achieved, as determined by densitometry analysis.
Purification of the pET28a-my32/Ls plasmid DNA
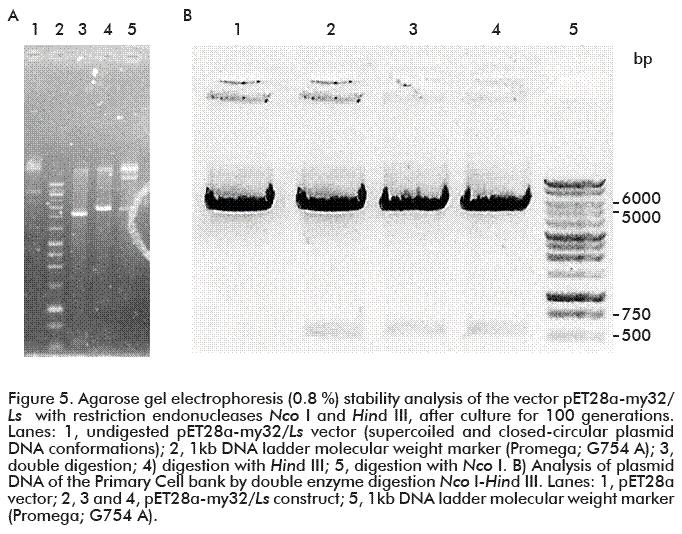
The pET28a-my32/Ls plasmid DNA was purified to analyze plasmid stability after 100 generations. The identity of the construct was checked by 0.8 % agarose gel electrophoresis followed by restriction analysis with Nco I and Hind III, and the expected restriction pattern was obtained. As shown in figure 5A, a band consistent with the 535 bp size of the my32/Ls gene was exscinded from the vector backbone (approximately 5246 bp) by Nco I-Hind III digestion. This pattern coincided with that of the PCB analysis (Figura 5B). The linear vector after the Hind III restriction corresponds to a band of approximately 5.7 kb (Figure 5A, lane 4). The preservation of the restriction pattern after 100 generations provided evidence of the preservation of plasmid integrity, what have to be thoroughly corroborated by sequencing.
Sequencing of the my32/Ls gene
After sequencing, a 100 % identity match was found between the established sequence for the gene my32/ Ls and that determined in plasmid samples taken after 100 cell generations of the E. coli BL21DE3-pET28a-my32/Ls strain.
DISCUSSION
Evaluating the genetic stability of the pET28a-my32/Ls gene construct in the protein production process during fermentation that double cell generations of the E. coli BL21(DE3) host cells
In this work, we demonstrate that the number of cell generations did not affected the genetic stability of the expression construct pET28a-my32/Ls in the PCB of E. coli BL21(DE3) transformed host cells. The genetic stability analysis provides information on the preservation of the genetic sequence of the PCB coding for the expression product, required to be stable to the end of the process. Besides, genetic stability is influenced by many factors, including the metabolic burden of the plasmid, copy number, replication species, type of growth substrate, medium composition, culture conditions, and the successive culture rounds [8].
For instance, Ashby and Stacey [17] reported that a plasmid-free bacterial strain showed a specific growth rate faster than the same strain carrying an expression plasmid, due to the increased metabolic energy demand for plasmid maintenance and replication within the cell. In our study, experimental data showed no statistical differences in the cell growth speed and cell generation doubling time of the strain during C1 and C3 propagation cycles, indicative of similar growth rates both in the presence and absence of the plasmid until the end of the fermentation process. Silva et al. [18] reported that plasmid instability could be caused by low growth rates in plasmid-carrying cells as compared to plasmid-free cells.
As could be inferred from the abovementioned aspects, plasmids are not always stable in cells culture for many generations in large-scale cultures. Therefore, it demands to study plasmid stability when changing the fermentation scale [10]. However, it could be possible that the sole change of fermentation scale does not always affect the plasmid genetic stability, since we found no changes in this parameter for the plasmid genetic construct under study. In fact, the pET28a-my32/Ls was shown to be quite stable after 100 generations. Similar results have also been obtained by other groups [8], also in E. coli BL21(DE3), at that time transformed with the pED-GnRH3-MVP construct expressing the gonadotropin-releasing hormone (GnRH) as a fusion protein under the control of the plac promoter, which was stable for up to 50 generations.
Furthermore, the level of expression achieved after 100 generations for the MY32/Ls provided further evidence of the stability and the expression efficiency of the pET28a vector backbone. The design of the expression process at the technological development scale, specifically at the end of the fermentation process, supported expression levels higher than 40 % after its induction by adding IPTG. The expression levels achieved in our study also confirm that the pET expression system is adequate to clone and express recombinant proteins in E. coli at industrial scale. In fact, the expressed protein could reach up to 50 % of the biomass total proteins just a few hours after induction [19]. In our case 56.9 % of total proteins corresponded to the MY32/Ls recombinant protein, similar to those obtained while checking the expression levels of the PCB (55.8 %, average expression value). Besides, bands in the range of 20-28 kDa were identified in SDS-PAGE, corresponding to the molecular weight expected for the MY32/Ls protein. Similar results were reported by Xu et al. [8], who found bands for the GnRH fusion protein at the expected size (> 20 kDa) after 4 h of induction with lactose. They also found expression levels higher than 40 % of total proteins in cultures after 50 generations.
The identity of the my32/Ls gene inserted into the pET28a plasmid harbored by the E. coli BL21(DE3)-pET28a-my32/Ls strain was preserved 100 % for 100 generations, as it was corroborated by sequencing, and the gene sequence completely matched that reported for the Primary Cell Bank. This was also indicative of the stability of the gene construct, which was devoid of gene modifications resulting from gene instability processes that affect expression vectors, such as insertions, direct or inverted repeats and deletions.
In summary, this study provided data confirming that the expression system for the production of the MY32/Ls protein is genetically stable during the entire production process in the host system used. This supports the generation of a working cell bank guaranteeing the stability, integrity and safety of the production process at large scale.
CONFLICTS OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
REFERENCES
1. Costello MJ. The global economic cost of sea lice to the salmonid farming industry. J Fish Dis. 2009;32(1):115-8.
2. Bravo S. The reproductive output of sea lice Caligus rogercresseyi under controlled conditions. Exp Parasitol. 2010;125(1):51-4.
3. Carpio Y, Basabe L, Acosta J, Rodriguez A, Mendoza A, Lisperger A, et al. Novel gene isolated from Caligus rogercresseyi: a promising target for vaccine development against sea lice. Vaccine. 2011;29(15):2810-20.
4. Ruiz LM, Orduz S, Lopez ED, Guzman F, Patarroyo ME, Armengol G. Immune response in mice and cattle after immunization with a Boophilus microplus DNA vaccine containing bm86 gene. Vet Parasitol. 2007;144(1-2):138-45.
5. Carpio Y, Garcia C, Pons T, Haussmann D, Rodriguez-Ramos T, Basabe L, et al. Akirins in sea lice: first steps towards a deeper understanding. Exp Parasitol. 2013;135(2):188-99.
6. Poutou RA, Amador E, Candelario M. Banco de Células Primario (BCP): Caracterización y papel en la producción de proteínas recombinantes. Biotecnol Apl. 1993;11(1)55-62.
7. Ramírez R, Muné M, Mansur M, Soto Y, Pimentel T. Estudio de estabilidad genética del sistema de expresión de la proteína E del virus dengue 4 en la levadura metilotrófica Pichia pastoris. Rev Cubana Med Trop. 2005;57:3.
8. Xu J, Li W, Wu J, Zhang Y, Zhu Z, Liu J, et al. Stability of plasmid and expression of a recombinant gonadotropin-releasing hormone (GnRH) vaccine in Escherichia coli. Appl Microbiol Biotechnol. 2006;73(4):780-8.
9. Fakruddin M, Mohammad Mazumdar R, Bin Mannan KS, Chowdhury A, Hossain MN. Critical factors affecting the success of cloning, expression, and mass production of enzymes by recombinant E. coli. ISRN Biotechnol. 2013;2013:590587.
10. Pierce J, Gutteridge S. Large-scale preparation of ribulosebisphosphate carboxylase from a recombinant system in Escherichia coli characterized by extreme plasmid instability. Appl Environ Microbiol. 1985;49(5):1094-100.
11. Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207-34.
12. Martínez J, Romay Z, Rojas T, Guerra G. Manual práctico de microbiología. La Habana: Ed. Pueblo y Educación; 1985.
13. Lee SY, Chang HN. High cell density cultivation of Escherichia coli W using sucrose as a carbon source. Biotechnol Lett. 1993;15:971-4.
14. Madigan M, Martinko JM, Parker J. Brock Microbiología de los microorganismos. Madrid: Pearson Educación; 2003.
15. Amador E, Almazan M, Quintana M, Poutou RA, Candelario M. Estudio preliminar de la estabilidad de los Bancos de Células Primarios para la producción de interferón alfa recombinante. Biotecnol Apl. 1994;11(1):60-3.
16. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680-5.
17. Ashby RE, Stacey KA. Stability of a plasmid F Trim in populations of a recombination-deficient strain of Escherichia coli in continuous culture. Antonie Van Leeuwenhoek. 1984;50(2):125-34.
18. Silva F, Lourenço O, Maia C, Queiroz JA, Domingues FC. Impact of plasmid induction strategy on overall plasmid DNA yield and E. coli physiology using flow cytometry and real-time PCR. Process Biochem. 2011;46:174-81.
19. Palomares LA, Estrada-Mondaca S, Ramirez OT. Production of recombinant proteins: challenges and solutions. Methods Mol Biol. 2004;267:15-52.
Received in May, 2017.
Accepted in October, 2017.
Niuvis Montoya. Centro de Ingeniería Genética y Biotecnología de Camagüey. Circunvalación Norte y Ave. Finlay, Camagüey, Camagüey, Cuba. E-mail: niuvis.montoya@cigb.edu.cu.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}