










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Marfan (MFS) es una enfermedad hereditaria congénita del tejido conectivo que se transmite con carácter autosómico dominante y penetrancia completa; su prevalencia se ha estimado en una por cada 5 000 personas, con una incidencia de uno por cada 10 000 nacimientos, sin predominio racial ni de sexo.
Se manifiesta en aquellos sistemas u órganos que contienen fibras elásticas en mayor concentración, tales como el sistema cardiovascular, el esquelético, la duramadre, los ojos, la piel, los tegumentos y los pulmones, aunque existen muchos casos de MFS parcial en los que el diagnóstico es dudoso y hay, además, gran variedad de cuadros clínicos asociados con las manifestaciones básicas de la enfermedad que ponen de manifiesto su heterogeneidad genética.1,2
MFS es un trastorno genético autosómico dominante que afecta al tejido conectivo, constituido por fibras elásticas que se compone de elastina y una red de microfibrillas ricas en fibrilina 1 codificada por el gen FBN1 en el cromosoma 15q21; el MFS es causado por una mutación genética FBN1 que altera la síntesis de fibrilina 1 y conduciendo a la debilidad del tejido, aumento de TGF-β, pérdida de las interacciones célula-matriz, lo que da lugar a las diferentes manifestaciones fenotípicas.3,4,5
La nosología de Ghent ha propuesto criterios clínicos para diagnosticar de manera oportuna esta patología, además de la aplicación de estrategias de manejo como evaluaciones periódicas, modificación de factores de riesgo, consejería genética y cirugía en ciertos pacientes. El propósito de este caso clínico es aportar aspectos actuales del MFS, que requiere una atención multidisciplinaria para obtener una disminución de la morbimortalidad.6,7
Presentación del caso
Paciente masculino de 8 años, talla 133 cm, peso 24 kg.
Antecedentes patológicos personales: disnea.
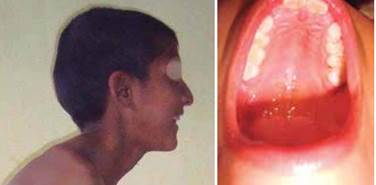
Examen físico: dolicostenomelia (extremidades elongadas en relación a las dimensiones del tronco), escaso panículo adiposo, gran envergadura, asimetría pectoral, como se muestra en la Figura 1. También se notó dolicocefalia y paladar ojival (Figura 2). A la auscultación se encontró soplo corto en foco aórtico accesorio.

Fig. 1 Dolicostenomelia (extremidades elongadas en relación a las dimensiones del tronco), escaso panículo adiposo, gran envergadura, asimetría pectoral
Se realizó ecocardiograma transtorácico que reportó dilatación. Los pediatras consultados no encontraron patologías; el genetista al encontrar mutación de Marfan solicitó examinar a los miembros de la familia, y el servicio de Ortopedia indicó una serie de estudios imagenológicos.
Se aplicaron los criterios de Ghent revisados, siguiendo el protocolo de evaluación diagnóstica se encontraron los siguientes datos:
Antecedentes patológicos familiares:
Padre de 32 años (Figura 3), diagnosticado de MFS en su juventud (Cardiología: diámetro aumentado de raíz aórtica de 4,6 cm, con 55 % de fracción de eyección, soplo diastólico en foco aórtico. Neumología: antecedente de tuberculosis pulmonar, deformidad torácica con crecimiento de cartílagos esternocostales derecho, se ha realizado controles seriados de tomografía axial computarizada y mostró bula que ha evolucionado y desplazó estructuras vecinas; presentó fibrosis cicatricial bilateral severa, además, de reporte espirométrico con patrón restrictivo severo. Ortopedia: escoliosis).

Fig. 3 Padre. 3A y 3B. Dolicostenomelia, dificultad para extensión de codo, asimetría de tórax (pectus carinatum). Aracnodactilia (falanges anormalmente largas), Escoliosis con presencia de bula, fibrosis cicatricial postuberculosis.
Luego de unos meses la disnea en el niño progresó, por lo que se transfirió al servicio de Cardiología Pediátrica.
Se detectó un soplo sistólico en foco mitral y se realizó un segundo ecocardiograma, que evidenció calcificación de la aorta con dilatación de su raíz y de la aorta descendente; se procedió a iniciar tratamiento farmacológico con losartán 12,5 mg cada 24 horas y propanolol 10 mg cada 24 horas, se logró mejorar el cuadro clínico.
Se identificaron las características fenotípicas de este paciente, así como sus hallazgos sugerentes de MFS; se decidió realizar interconsulta multidisciplinaria (Oftalmología: no reportó alteraciones estructurales).
Madre del paciente tiene estatura promedio sin fenotipo marfanoide.
Tío paterno del paciente inicial con MFS falleció hace 5 meses por disección aórtica.
El paciente tiene un primo de 7 años, de estatura alta para su edad, presentó hiperlaxitud articular y pie bilateral plano, además, tiene una hermana menor que está sana, con un desarrollo psicomotor y exploración normales.
Las pruebas genéticas se usaron para confirmar el diagnóstico de síndrome de Marfan, como en este caso presentado.
Discusión del caso
Dado el impacto del pronóstico y el manejo, la terapia médica temprana y la intervención quirúrgica oportuna en caso que lo amerite ha mejorado sustancialmente la calidad de vida comparada con décadas anteriores en esta enfermedad.
La evaluación diagnóstica del MFS es inevitablemente compleja, la falla en su detección puede estar dada debido a la marcada variabilidad de presentación (intra e interfamiliar), en dependencia de la edad en la aparición de manifestaciones clínicas, ausencia de la técnica diagnóstica que define la presencia de la condición con la máxima certeza conocida, amplios diagnósticos diferenciales y la existencia de mutaciones.8,9,10
Actualmente, los pilares fundamentales de la terapéutica farmacológica son los betabloqueantes y antagonistas del receptor de angiotensina, pues tienen la propiedad de disminuir el inotropismo, frecuencia cardíaca, presión arterial y rigidez aórtica, es decir, reducen la frecuencia de complicaciones cardiovasculares.
En el caso reportado, con el diagnóstico precoz, tratamiento y seguimiento, se estimó obtenga una amplia expectativa de vida, es decir, tendrá un pronóstico bueno; sin embargo, el padre, debido a las características clínica y tardío manejo, es un paciente de alto riesgo para cirugía y deberá evaluarse la necesidad quirúrgica.11
Consideraciones finales
El síndrome de Marfan es una patología poco común, causada por una mutación genética de fibrilina 1, imprescindible para la síntesis de fibras elásticas del tejido conectivo.
La toma de decisiones es compleja, debe iniciarse tempranamente su manejo multidisciplinario con un seguimiento minucioso; el diagnóstico requiere una evaluación clínica completa de múltiples órganos y sistemas, por su ampliada sintomatología, ya que se dispone de tratamientos farmacológicos y quirúrgicos que mejoran la esperanza de vida.

