










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMEDISAN
On-line version ISSN 1029-3019
MEDISAN vol.17 no.4 Santiago de Cuba Apr. 2013
CASO CLÍNICO
Aneurismas de aorta abdominal e ilíaca y síndrome de Loeys- Dietz
Abdominal aortic iliac aneurysms and Loeys-Dietz syndrome
MsC. David Ortiz Limonta, MsC. Dalia Sánchez De la Guardia y MsC. Lilia Chércoles Cazate
Hospital Provincial Docente Clinicoquirúrgico "Saturnino Lora Torres", Santiago de Cuba, Cuba.
RESUMEN
Se presenta el caso clínico de una paciente de 29 años de edad, con antecedentes de ectasia aórtica, quien había sido estudiada a los 18 años mediante cineangiografía y se halló un aneurisma fusiforme de la aorta abdominal infrarrenal y de la arteria ilíaca primitiva izquierda, de causa no precisada, las cuales fueron sustituidas con derivación aortofemoral izquierda. Posteriormente, en el 2012, presentó dolor y aumento del volumen en la región inguinal izquierda, por lo que fue atendida en el Cuerpo de Guardia del Hospital Provincial Docente Clinicoquirúrgico "Saturnino Lora Torres" de Santiago de Cuba, y evaluada por especialistas del Servicio de Angiología y Cirugía Vascular, los cuales determinaron operarle de urgencia, al observar un aneurisma anastomótico de la arteria femoral izquierda, de causa micótica, y las características: craneosinostosis, úvula bífida y hendidura del paladar; propias del síndrome de Loeys-Dietz.
Palabras clave: síndrome de Loeys-Dietz, aneurisma de la aorta abdominal, aneurisma anastomótico de la arteria femoral, derivación aortofemoral izquierda, Servicio de Angiología y Cirugía Vascular.
ABSTRACT
The case report of a 29 year-old patient, with a history of aortic ectasia who had been studied when she was 18 years through cineangiography is presented and a fusiform aneurysm of the infrarrenal abdominal aorta and of the left primitive iliac artery, of unspecific cause was found, which were substituted with left aortofemoral bypass. Later on, in 2012, she presented pain and increase of the volume in the left inguinal region, reason why she was assisted in the emergency room of "Saturnino Lora Torres" Teaching Provincial Clinical Surgical Hospital in Santiago de Cuba, and evaluated by specialists of the Angiology and Vascular Surgery Service, which determined to treat her with an emergency surgery, when observing an anastomotic aneurysm of the left femoral artery of fungal cause, and the characteristics: craneosinostosis, bifid uvula and cleft palate; characteristic of Loeys Dietz syndrome.
Key words: Loeys-Dietz syndrome, abdominal aorta aneurysm, anastomotic aneurysm of the femoral artery, left aortofemoral bypass, Angiology and Vascular Surgery Service.
INTRODUCCIÓN
El síndrome de Loeys-Dietz es una enfermedad autosómica dominante del tejido conectivo, que había sido referida previamente, en 1987, como síndrome de Furlong, y en el año 2005, fue descrita por Bart L. Loeys (Centro de Medicina Genética de Gante en Bélgica) y Harry Dietz (Facultad de Medicina de la Universidad Johns Hopkins de Baltimore), y adquirió el nombre actual.1,2
Inicialmente se estudiaron 10 familias y se obtuvieron los siguientes hallazgos clínicos:
- Manifestaciones vasculares: Dilatación o disección de la aorta (en 95 % de los afectados) y aneurisma arterial. Pueden existir prolapso valvular mitral, ectasia de la arteria pulmonar, ductus arterioso persistente, así como defectos septales atriales y válvula aórtica bicúspide.3
- Manifestaciones esqueléticas: Pectus excavatum o pectus carinatum, escoliosis, laxitud articular, signos del pulgar y la muñeca positivos, y pie equino varo. Suelen presentarse aracnodactilia, pies planos y osteoporosis.2,3
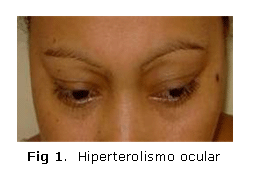
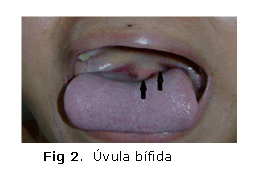
- Manifestaciones craneofaciales: Hipertelorismo ocular, úvula bífida, paladar hendido, craneosinostosis con dolicocefalia, braquicefalia. Pueden tener hipoplasia malar con retrognatia.2,3
- Manifestaciones cutáneas: Piel fina, friable, con circulación venosa muy visible y mala cicatrización.2,4
- Otras manifestaciones: Trastornos de refracción (miopía), displasia del esmalte dentario e inestabilidad atlantoaxil; también se ha notificado rotura espontánea del bazo, el intestino o el útero durante la gestación.3,4
Este síndrome se asocia a mutaciones del gen FBN-1, que codifica los receptores de tipo I (TGFBR1) y tipo II (TGFBR2) del factor de crecimiento de transformación beta (TGF-B). Las mutaciones producen un aumento de la señalización intracelular del TGF-B, y ello da lugar a un aumento en la producción de metaloproteinasas (MMP-2 y MMP-9) y de ácido hialurónico en el espacio extracelular. El resultado es la incapacidad del tejido conectivo para mantenerse unido, lo que constituye una amenaza importante para la integridad de la aorta y de muchos vasos sanguíneos.2,5,6
Una de las dificultades para detectar la enfermedad es que puede ser confundida con el síndrome de Marfan, aunque posee un peor pronóstico, pues los pacientes mueren con una edad media de 20 a 22 años. Al respecto, la afectación arterial, en especial la dilatación aórtica y la disección, es la causa más frecuente de muerte.6, 7
Dado que la presentación de los aneurismas de la aorta abdominal y el pronóstico de su rotura, son infrecuentes en estas edades de la vida, es de vital importancia el diagnóstico temprano y el tratamiento oportuno de enfermedades que puedan agravar dicha entidad clínica. Por tal motivo, se decidió presentar este trabajo, que puede resultar de interés para angiólogos, cirujanos vasculares y cirujanos generales.
CASO CLÍNICO
Se describe el caso clínico de una paciente de 29 años de edad, con antecedentes de un aneurisma de la aorta abdominal infrarrenal y de la arteria ilíaca primitiva izquierda, de causa no precisada, a la edad de 18 años, las cuales fueron sustituidas con derivación aortofemoral izquierda, por una prótesis de dacrón del tipo punto de malla (14 x 7 mm de diámetro), en forma bifurcada.
Luego egresó y, por una decisión personal, no mantuvo el seguimiento clínico en la consulta de Cirugía Vascular, durante 11 años. En ese tiempo la paciente tuvo una vida normal, que incluyó un embarazo a término con parto transpelviano (2 años antes), y un legrado uterino para interrumpir otra gestación, hacía solo 2 semanas.
El 3 de septiembre del 2012 fue remitida, desde su área de salud, al Cuerpo de Guardia del Hospital Provincial Docente Clinicoquirúrgico "Saturnino Lora Torres" de Santiago de Cuba, por presentar dolor y aumento del volumen de la región inguinal izquierda. Fue evaluada por el especialista en Angiología y Cirugía Vascular de guardia, quien además tuvo en cuenta la operación realizada años atrás, que contaba en los registros quirúrgicos del Servicio, así como la falta de seguimiento clínico ambulatorio, debido a una decisión personal de la paciente y sus familiares, bajo el argumento de haberse sentido bien hasta ese momento. La paciente no refería claudicación intermitente durante la marcha.
Examen físico
En el sistema arterial periférico de sus extremidades inferiores presentaba los pulsos femoral, poplíteo, tibial posterior y pedio derechos; sin embargo, no había pulso alguno en el miembro inferior izquierdo. De igual manera, se detectó un tumor inguinal en el lado izquierdo, de aproximadamente 10 a 15 centímetros, debajo de la cicatriz producida por la intervención quirúrgica, el cual no tenía latidos ni se dilataba, ni presentaba soplo; tampoco había signos de isquemia en la extremidad, pero sí se observaron ligeras hipertermia y eritema de la piel.
Se indicó una tomografía computarizada multicorte, contrastada, de urgencia, con el diagnóstico presuntivo de sustitución de la aorta y la arteria ilíaca derechas, como consecuencia de una complicación en la derivación aortofemoral izquierda debido a una trombosis, y posible rotura de aneurisma anastomótico en la prótesis.
También se observó que la paciente presentaba signos físicos característicos como hipertelorismo ocular (figura 1) y úvula bífida (figura 2) o hendidura del paladar, que definen el diagnóstico clínico del síndrome de Loeys-Dietz.
Estudios realizados
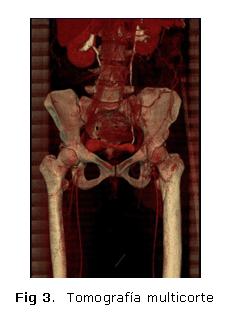
La angiografía, mediante el tomógrafo multicorte, posibilitó visualizar la sustitución de la aorta infrarrenal y la arteria ilíaca derechas, con trombosis antigua de la rama izquierda en la derivación aortofemoral. Se observó, además, una gruesa circulación colateral creada desde el segmento de la aorta yuxtarrenal, que descendía en derivación fisiológica hasta la aorta femoral superficial del lado izquierdo y llenaba parte del tumor inguinal de ese lado (figura 3), así como las regiones de las arterias femorales superficial y profunda, y la zona poplítea.
Tratamiento quirúrgico
Se decidió intervenir quirúrgicamente con urgencia ante el diagnóstico de aneurisma anastomótico, en la unión de la prótesis y la arteria femoral izquierda, además de una trombosis en la zona de la derivación.
La incisión se realizó en "palo de jockey", por vía extraperitoneal, hasta la rama izquierda de la prótesis en derivación (con trombosis y sin pulso), la cual fue cargada y del conducto fluyó, desde la arteria femoral hacia el retroperitoneo, una secreción purulenta y fétida, que era parte del contenido del tumor inguinal. Se descartó la posibilidad de revascularizar, y se decidió extraer la rama izquierda de la prótesis a causa de la sepsis y la trombosis.
Al exponer la región inguinal se halló un aneurisma micótico y anastomótico en la prótesis de la arteria femoral, con aproximadamente 500 mililitros de secreción purulenta y fétida, y un buen flujo que retrocedía en las arterias femorales (superficial y profunda). Se logró la hemostasia de ambas arterias con el uso de las pinzas de Satinsky; asimismo, se extrajo la prótesis desde el retroperitoneo a través de la región inguinal izquierda, y se dejó un tramo de drenaje en el conducto, que salía por la parte interna del muslo.
Posteriormente se realizó endoaneurismorrafia en el orificio anastomótico de la arteria femoral común (profunda y superficial), para lo cual se empleó sutura monofilamento, del tipo polipropileno 3 x 0, sin afectar la derivación fisiológica que llegaba a la arteria femoral superficial y salía de la aorta abdominal yuxtarrenal.
Se tomaron muestras de secreción para cultivo, y además se realizó hemocultivo. Se administró antibiótico endovenoso en el período peroperatorio (ceftriaxona de 1 gramo). La extremidad izquierda quedó sin pulso, aunque tampoco presentaba signos de isquemia; era viable a través de la derivación fisiológica creada, la cual no fue dañada en el procedimiento quirúrgico.
En el período posoperatorio se administró una tríada de antimicrobianos: meropenem, metronidazol y amikacina, durante los 10 primeros días, y posteriormente se aplicó Trifamox®. Luego la paciente fue tratada con cloranfenicol (1 gramo) por vía endovenosa cada 12 horas, pues el resultado del cultivo y antibiograma mostró la presencia de un tipo de klebsiella con sensibilidad al meropenem, ciprofloxacina, cefazolina, ceftriaxona, cloranfenicol, kanamicina, amikacina y gentamicina.
Por otra parte, el hemocultivo dio negativo. No hubo crecimiento de microorganismos.
La paciente fue egresada a los 30 días y se le indicó tratamiento ambulatorio con ciprofloxacina (250 mg), 2 tabletas cada 12 horas, ozonoterapia por vía rectal en 15 sesiones y seguimiento clínico cada mes en la consulta de Cirugía Vascular, con el protocolo de ciclos profilácticos mensuales a base de antimicrobianos y la repetición de la ozonoterapia cada 6 meses.
COMENTARIOS
Desde el punto de vista clínico, se han definido 3 características que distinguen al síndrome de Loeys-Dietz: tortuosidad excesiva y aneurismas arteriales, hipertelorismo, y úvula bífida o hendidura del paladar. Esta descripción se corresponde con lo hallado en la paciente de este caso clínico, lo cual condujo a determinar el diagnóstico clínico de la enfermedad.6,8
Recientemente, dicho síndrome ha sido clasificado en tipo I, cuando existe afectación craneofacial (hendidura del paladar, hipertelorismo o craneosinostosis), y tipo II, si no hay tal afectación, pero se observan úvula bífida y predominio de los signos presentes en el síndrome de Ehlers-Danlos, al menos 2 de ellos (rotura visceral, laxitud articular, piel traslúcida o aterciopelada, dermatólisis, cicatrización amplia y atrófica).5,9
Aquellos pacientes con afectación craneofacial, tienen habitualmente un daño arterial más grave y "agresivo", lo cual coincide con loas manifestaciones de la paciente de este artículo, quien presentó el tipo I de la enfermedad, con afectación masiva de todo el árbol arterial, evidenciada a través de los estudios de imágenes. Los individuos que padecen la enfermedad en su tipo I, necesitan una operación precoz, con una media de edad de 16,9 años, respecto a los que presentan el tipo II, que tienen una media de 26,9 años, y mueren mucho antes (22,6 años versus 31,8 en el tipo II).2
La dilatación o disección aórtica, o ambas, tienen lugar en edades más tempranas y con diámetros arteriales menores, en comparación con las características del síndrome de Marfan; por ello, es necesario un diagnóstico y tratamiento quirúrgico precoces, si el diámetro de la aorta excede a 40 mm.1,9
REFERENCIAS BIBLIOGRÁFICAS
1. Furlong J, Kurczynski TW, Hennessy JR. New Marfanoid syndrome with craniosynostosis. Am J Med Genet. 1987; 26(3): 599-604.
2. Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005; 37(3): 273-81.
3. Loeys BL, Dietz HC. Loeys-Dietz Syndrome. En: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP. Gene Reviews. Seattle: University of Washington; 1993.
4. Yetman AT, Beroukhim RS, Ivy DD, Manchester D. Importance of the clinical recognition of Loeys-Dietz syndrome in the neonatal period. Pediatrics. 2007; 119: 1199-202.
5. Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutationsin the TGF-beta receptor. New Eng J Med. 2006; 355(8): 788-98.
6. Singh KK, Rommel K, Mishra A, Karck M, Haverich A, Schmidtke J, et al. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum Mutat. 2006; 27(8): 770-77.
7. Viassolo V, Lituania M, Marasini M, Dietz H, Benelli F, Forzano F, et al. Fetal aortic root dilation: A prenatal feature of the Loeys-Dietz syndrome. Prenat Diagn. 2006; 26(11): 1081-83.
8. Akutsu K, Morisaki H, Takeshita S, Sakamoto SH, Tamori Y, Yoshimuta T, et al. Phenotypic heterogeneity of Marfan-like connective tissue disorders associated with mutations in the transforming growth factor-receptor genes. Circ J. 2007; 71(8): 1305-09.
9. Spin JM. Gene mutations and familial thoracic aortic aneurysms: a walk on the mild side. Circ Cardiovasc Genet. 2011; 4(1): 4-6.
Recibido:11 de febrero de 2013.
Aprobado: 20 de marzo de 2013.
David Ortiz Limonta. Hospital Provincial Docente Clinicoquirúrgico "Saturnino Lora Torres", avenida de los Libertadores s/n, entre calles 4ta y 6ta, reparto Sueño, Santiago de Cuba, Cuba. Correo electrónico:dortiz@medired.scu.sld.cu
