










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
On-line version ISSN 1561-3194
Rev Ciencias Médicas vol.17 no.4 Pinar del Río July-Aug. 2013
ARTÍCULO ORIGINAL
Caracterización del Síndrome de Down en la población pediátrica
Characterization of Down syndrome in Pediatric population
Odilkys Cala Hernández
Licenciada en Defectología. Master en Atención Integral al niño. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico: odigen@princesa.pri.sld.cu
RESUMEN
Introducción: el Síndrome de Down es una enfermedad genética que constituye la primera causa de retraso mental. El incremento de la calidad de vida en la población, el desarrollo de herramientas para el asesoramiento genético, el mejoramiento de los servicios de salud y educación, que incluyen la rehabilitación, motivan al continuo esfuerzo por el dominio de la enfermedad.
Objetivo: caracterizar clínica-epidemiológicamente el Síndrome de Down en la población pediátrica de Pinar del Río.
Material y método: se realizó un estudio descriptivo, longitudinal en pacientes con diagnóstico de Síndrome de Down atendidos en el Centro Provincial de Genética Médica de Pinar del Río. La muestra estuvo constituida por 110 pacientes menores de 17 años, 11 meses y 29 días. Se revisaron las historias clínicas y se realizó un consentimiento informado.
Resultados: predominaron los pacientes del sexo femenino, la edad materna de mayor riesgo para la presentación del Síndrome de Down fue el grupo de edades comprendido entre 31-35 años. Los defectos congénitos cardiovasculares que se presentaron con mayor frecuencia fueron la comunicación interventricular y comunicación interauricular. El mayor porciento de los casos con cariotipo anormal correspondió a trisomía libre.
Conclusiones: con el estudio clínico y epidemiológico se logra un mejor manejo de las secuelas discapacitantes de la enfermedad, y se aportan datos científicos necesarios para el asesoramiento genético. Se realizó un estudio en 110 pacientes menores de 18 años, atendidos en el Centro Provincial de Genética Médica en la ciudad de Pinar del Río (Cuba) durante los años 1993-2011.
DeCS: Síndrome de Down, Familia, Enfermedades genéticas congénitas.
ABSTRACT
Introduction: Down syndrome is a genetic disease that constitutes the first cause of mental retardation. The increase of the quality of life in the population, the development of equipments to the carry out the genetic counselling, the improvement of health services and education which includes the rehabilitation, all these motivate the continuing effort to control the disease.
Objective: to characterize Down syndrome from the clinical and epidemiological point of view in pediatric population, Pinar del Rio.
Material and Method: a descriptive, longitudinal study was carried out in patients with the diagnosis of Down syndrome and attending at Provincial Center of Medical Genetics, Pinar del Rio. The sample included 110 patients younger than 17 years old, 11 month and 29 days. Clinical histories were reviewed and an informed consent was applied.
Results: female sex prevailed; maternal ages from 31 to 35 were in risk for Down syndrome presentation. Ventricular and Atrial septal defects were the most frequent cardiovascular congenital defects. The greatest percentage of cases with abnormal karyotype belonged to a free trisomy.
Conclusions: by means of a clinical and epidemiological study a better management of disabling sequels of the disease is achieved, along with scientific necessary data to carry out the genetic counselling. The study comprised 110 patients younger than 18 years old who attended at Provincial Center of Medical Genetics in Pinar del Rio (Cuba) all along 1993-2011.
DeCS: Down Síndrome, Family, Inborn genetic diseases.
INTRODUCCIÓN
En la actualidad se considera que un 10% de la población mundial, representado por 650 millones de personas, viven con alguna ausencia de estado óptimo (discapacidad) (Organización de las Naciones Unidas, "ONU"). 1 Se estima que esta cifra seguirá aumentando, debido al crecimiento y envejecimiento de la población. Por otra parte, las estadísticas del Banco Mundial (2010) estiman que el 20% de los más pobres en el mundo presentan algún tipo de discapacidad y tienden a ser considerados dentro de sus propias comunidades como las personas en situación más desventajosa.1,2
El devenir histórico de la humanidad reconoce que el destino de las personas con discapacidad ha dependido de las actitudes y comportamiento prevalentes entre las personas que disfrutan de un estado óptimo de vida, signado por las diferentes organizaciones sociales.
Nuestro país se identifica con un proyecto social que tiene como finalidad la prosperidad, la independencia, el desarrollo humano sostenible y la preservación de la identidad cultural, contemplando la integración de todos sus miembros, lo que ha constituido brújula para la orientación de la toma de decisiones en la atención a personas en general y discapacitados en particular. En consecuencia, entre los años 2001-2003, en el marco de la batalla de ideas que libra nuestro pueblo, se realizó un estudio psicosocial que permitió la evaluación de todos los grupos de edades con alguna ausencia de estado óptimo.3 Se mantiene esta línea de trabajo como elemento indispensable en el funcionamiento de nuestras instituciones, partiendo de la formación de valores para lograr en realidad la atención a la diversidad que se presenta, y también forma parte indisoluble de los objetivos de trabajo e indicadores a cumplir por el Ministerio de Salud Pública, donde se plantea como número uno: Incrementar el estado de salud de la población y su satisfacción con los servicios, precisándose la necesidad de mantener actualizados los registros de discapacitados y brindar atención a sus principales necesidades.4
Así las indicaciones realizadas por el Ministerio de Salud Pública presuponen un cambio de concepción hacia una posición más humanista que trasciende en la atención, no solo desde una posición clínica ¨clasificatoria¨, sino que se distingue por la atención integral, que presupone a su vez la inclusión de la familia. Se tiene en consideración que el hogar constituye la primera escuela para los niños, y sus padres, los primeros maestros (Unicef, 1992). 3,4
El Dr. John Longdon Down descubre que el Síndrome de Down es un conjunto de signos y síntomas presentes en una persona, provocados por una alteración genética a nivel del cromosoma 21, que determina en un ser humano una serie de características fenotípicas propias de este padecimiento, convirtiéndose en la primera causa de retardo mental de origen genético.5
Como es conocido, enfrentar en una familia el reto de un bebé que presenta características diferentes a las que se esperan, provoca una conmoción entre todos sus miembros. Sin embargo, no siempre el personal de salud cuenta con los argumentos necesarios para orientar a la familia, en función de convertirlos en potenciadores de desarrollo del niño discapacitado.
En Pinar del Río se realizó un estudio exploratorio donde se pudo comprobar que aún cuando se cuenta con un registro de discapacitados, no se precisan las características de esta población de modo que se intervenga en virtud de potenciar su desarrollo dentro de la sociedad, a partir de la atención integral que involucre a la familia. De igual forma, se comprobó que no existe un material orientador.
Nuestro trabajo está dirigido hacia la elaboración de un estudio clínico y epidemiológico que permita al personal de salud en los servicios de genética médica, manejar herramientas que le permitan brindar la orientación necesaria a la familia y los pacientes con Síndrome de Down, con la intención de que se fortalezca tanto la atención al niño como a los que le rodean; y estará encaminada a proponer los pasos a seguir desde el momento mismo del nacimiento.
MATERIAL Y MÉTODO
Se realizó un estudio descriptivo, longitudinal, en un grupo de pacientes diagnosticados con Síndrome de Down, atendidos en el Centro Provincial de Genética Médica menores de 17 años, 11 meses y 29 días. El universo de este trabajo estuvo constituido por el total de pacientes con Síndrome de Down, 110 niños, que han asistido a los servicios de genética clínica hasta diciembre del año 2011, estudiándose clínicamente en su totalidad y lo anterior coincide con la muestra.
La selección de la muestra se realizó por un muestreo estratificado, teniendo en cuenta criterios de inclusión y exclusión. Todos los pacientes fueorn menores de 17 años, 11 meses y 29 días, pertenecían a la provincia Pinar del Río y sus padres dieron el consentimiento informado.
No se tomaron en consideración para la investigación los pacientes que no pertenecían a la provincia Pinar del Río, y a los pacientes que fallecieron en el período de estudio.
Los resultados se presentaron en tablas. Los resultados del análisis de las variables se resumieron en frecuencias absolutas y por cientos. Para esta investigación se solicitó a los pacientes el consentimiento informado, explicándoles la importancia de esta investigación y los beneficios que se pueden obtener. Se tuvieron en cuenta los fundamentos éticos del asesoramiento genético.
RESULTADOS
La muestra es de 110 pacientes (Tabla 1). 57 son del sexo femenino para un 51.8%, y 53 son del sexo masculino para un 48.2%.
Respecto a la edad de los progenitores, es necesario destacar que el riesgo de niños con Síndrome de Down se incrementa a medida que avanza la edad materna y paterna, (Tabla 2).
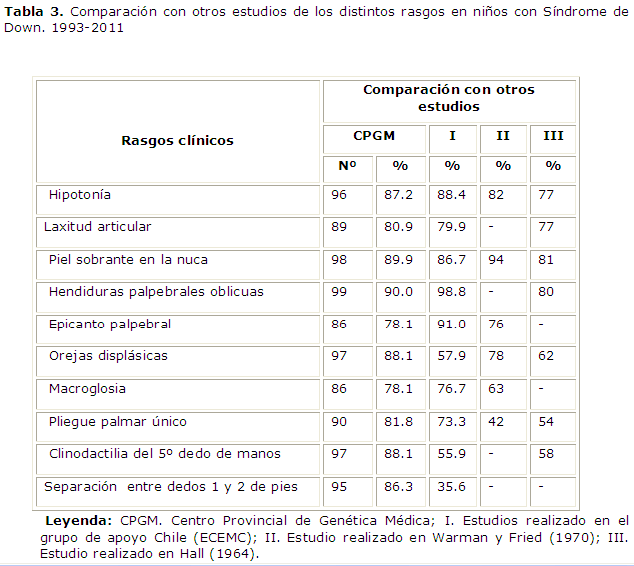
El rango de edades con mayor número de nacimientos fue de 31-35 años para la edad materna (28.4%), este estudio se corresponde con lo citado en la literatura, según la cual la edad media materna es de 34 años. (Tabla 3).
Como se puede apreciar, no todos los estudios incluyen los mismos rasgos fenotipicos. De hecho, algunos autores incluyeron 10 rasgos, entre los que se encontraban 7 de este estudio. Otros autores también seleccionan 10 rasgos, entre los que se encuentran 7 de esta investigación, y otros estudios incluyen 7 entre los 8 más frecuentes que se analizan. 4,5
En los defectos cardiovasculares en general se presenta la CIV (comunicación interventricular) como el defecto congénito más frecuente (gráfico), con un total de 20 casos para un 18.2 %, le sigue en orden decreciente en su frecuencia la CIV con CIA (comunicación interauricular), con 12 casos para un 10.9%.

En este estudio realizado, 44 casos presentan cardiopatía congénita, para un 40%, y las más frecuentes son la CIV y la CIA.
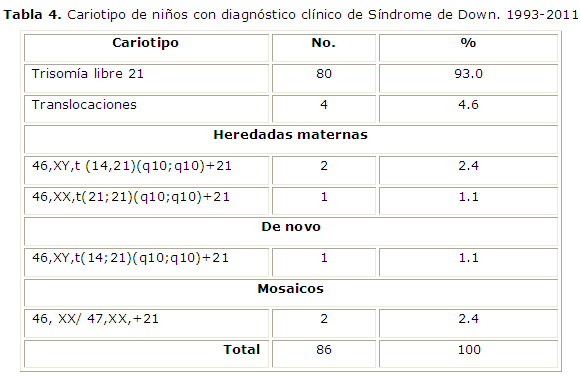
Se presentan los resultados de los estudios cromosómicos. De los 110 pacientes con diagnóstico clínico de Síndrome de Down, solo se le realizó estudio cromosómico a 86, al resto, que fueron 24, no se le realizó por diversas causas, como negación de los padres, entre otras.
El mayor por ciento de los casos con cariotipo anormal correspondió a trisomía libre, con 93 %. En este estudio se presentan 2 pacientes con diagnóstico cromosómico de mosaicismo, lo cual representa un 2.4 % de la muestra estudiada. En el caso de las translocaciones un 4.6 % involucra los cromosoma 14 y 21, con predominio en el sexo masculino. (Tabla 4).
DISCUSIÓN
Respecto a la edad de los progenitores, es necesario destacar que el riesgo de niños con Síndrome de Down se incrementa a medida que avanza la edad materna y paterna. El rango de edades con mayor número de nacimientos fue de 31-35 años para la edad materna, este estudio se corresponde con lo citado en la literatura, donde la edad media materna es de 34 años.
El aumento de la frecuencia de Síndrome de Down en el momento del nacimiento en relación con el aumento de la edad materna se ha documentado ampliamente. El mecanismo responsable de este efecto parece estar relacionado con el riesgo aumentado de no disyunción con la edad materna (hipótesis de la línea de producción). En otros estudios se ha analizado la frecuencia en el nacimiento de Síndrome de Down por años, 6 se encuentra una disminución de ésta, y una disminución de la frecuencia de Síndrome de Down en mujeres mayores de 34 años.
Se encontró que la frecuencia total de Síndrome de Down en mujeres de menos de 35 años va en aumento, y la edad media de la madre de un recién nacido con Síndrome de Down va disminuyendo, posiblemente por dos razones principales: la primera es el retraso en la edad materna de concepción, lo que produce un mayor número relativo de nacimientos en edades por encima de los 30 años, con mayor riesgo para las anomalías cromosómicas numéricas que en edades más jóvenes, riesgo que aumenta de manera progresiva con la edad materna. La segunda puede ser la generalización de la oferta de medios de diagnóstico prenatal invasivo (amniocentesis) a las gestantes mayores de 37 años en Cuba, lo que conlleva un aumento de las interrupciones voluntarias del embarazo en el grupo con mayor riesgo de aneuploidías, y en consecuencia a una disminución de recién nacidos con aneuploidía de madres mayores. Debido a que las medidas de diagnóstico prenatal invasivo no tienen una relación riesgo-beneficio adecuado por debajo de los 35 años, la disminución de la frecuencia de aberraciones cromosómicas por debajo de esta edad materna sólo puede conseguirse mediante la aplicación de medidas de cribado poblacional. Éstas incluyen los marcadores bioquímicos (alfafetoproteína, hormona gonadotropina coriónica, estriol, proteína plasmática A asociada al embarazo y los marcadores de ultrasonografía obstétrica incluyendo malformaciones mayores y "signos blandos" (pliegue nucal aumentado, colon hiperecogénico, fémur corto, foco ecogénico intracardíaco).7
Son numerosos los estudios que analizan la frecuencia de distintos rasgos clínicos entre los niños con Síndrome de Down. La identificación clínica del Síndrome de Down debe basarse en la ocurrencia en un mismo niño de un conjunto de rasgos y anomalías que individualmente no son específicas. Los resultados de este trabajo implican que los rasgos seleccionados tienen un alto valor predictivo para el diagnóstico de este síndrome. De hecho, sean cuales sean los rasgos que se seleccionen, el diagnóstico clínico no puede basarse en la presencia de uno solo de ellos, sino en la concurrencia de varios, que es en definitiva el patrón de signos dismórficos. La mayoría de los rasgos o signos dismórficos que se describen en el presente estudio han sido descritos por examen clínico en la etapa neonatal, y coinciden con los criterios de Hall en el neonato. 8-10
El Síndrome Down (SD) es la cromosomopatía más frecuente en humanos con incidencia de 1:700. El 40-60% de los SD tienen cardiopatías congénitas (CC), y son más frecuentes el canal atrio ventricular (AV), comunicación interventricular (CIV) y tetralogía de Fallot. 11 En este estudio realizado, 44 casos presentan CC, para un 40%, coincidiendo con la frecuencia reportada anteriormente, pero sin embargo las cardiopatías congénitas más frecuentes son la CIV y la CIA, esta última no es la más frecuentemente descrita por otros estudios. 12,13
Existen variaciones en la frecuencia de defectos congénitos cardiovasculares en estos pacientes en diferentes poblaciones estudiadas. Las variaciones en estos pacientes se atribuyen a factores genéticos y ambientales. Raras veces se reportan estenosis pulmonar valvular, atresia aórtica y coartación de la aorta, así como doble emergencia de ventrículo derecho, tronco arterioso y transposición de grandes vasos. 14
Las alteraciones cromosómicas son una de las principales causas de defectos congénitos mayores en recién nacidos. La presencia de un síndrome malformativo o dismórfico es una de las principales indicaciones de realización de un estudio citogenético. El conocimiento de la verdadera frecuencia de las aberraciones cromosómicas y de los aspectos epidemiológicos relacionados con éstas supone un importante avance en el estudio de la etiopatogenia de las malformaciones congénitas y de las cromosomopatías asociadas a ellas. El mayor por ciento de los casos con cariotipo anormal correspondió a trisomía libre, con 93 %, lo cual se corresponde con lo que se reporta en la literatura que algunos estudios reportan entre un 87 % y 95 %. En este estudio se reportan pacientes con Síndrome de Down secundario a mosaicismo cromosómico y translocación que involucra los cromosoma 14 y 21, con predominio en el sexo masculino; esto se corresponde con el estudio realizado en Chile, donde se reportan cifras cercanas a este estudio.15-16
Con el estudio epidemiológico y clínico de niños con Síndrome de Down se logra un mejor manejo de las secuelas discapacitantes de la enfermedad. Este estudio contribuye a aumentar los datos científicos sobre el comportamiento de esta entidad, con el propósito de aportar elementos necesarios para su diagnóstico, tratamiento y seguimiento, logrando un impacto sobre la calidad de vida.
REFERENCIAS BIBLIOGRÁFICAS
1. Lorenzi H, Duvall N, Cherry SM, Reeves RH, Roper RJ. PCR prescreen for genotyping the Ts65Dn mouse model of Down syndrome. Biotechniques [serie en Internet]. Ene 2010 [citado 5 Oct 2011]; 48(1): [aprox. 9 pantallas]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2955870/pdf/nihms240611.pdf
2. Boff J, Aquino Caregnato RC. História oral de mulheres com filhos portadores de Síndrome de Down. Texto & contexto enferm [serie en Internet]. Jul-Sep 2008 [citado 5 Oct 2011]; 17(3): [aprox. 9 pantallas]. Disponible en: http://www.scielo.br/pdf/tce/v17n3/a20v17n3.pdf
3. Cammarata-Scalisi F, Da Silva G, Cammarata-Scalisi G, Sifuentes A. Historia del Síndrome de Down. Un recuento lleno de protagonistas. Can Pediatr [serie en Internet]. 2010 [citado 5 Oct 2011]; 34(3): [aprox. 3 pantallas]. Disponible en: http://www.scptfe.com/inic/download.php?idfichero=533
4. Guedj F, Sébrié C, Rivals I, Ledru A, Paly E, Bizot JC, et al. Green Tea Polyphenols Rescue of Brain Defects Induced by Overexpression of DYRK1A. Plos ONE [serie en Internet]. Feb 2009 [citado 5 Oct 2011]; 4(2): [aprox. 8 pantallas]. Disponible en: http://www.plosone.org/article/fetchObjectAttachment.action;jsessionid=ED2B9E86B3705FF92D533EB70AE8DCB5?uri=info%3Adoi%2F10.
1371%2Fjournal.pone.0004606&representation=PDF.
5. Colectivo de autores. Por la vida. Estudio psicosocial de las personas con discapacidades y estudio psicopedagógico social y clínico-genético de las personas con retraso mental en Cuba. Ciudad de La Habana: Casa Editorial Abril,2003.
6. Alves-Sampaio A, Troca-Marín JA, Montesinos ML. NMDA-mediated regulation of DSCAM dendritic local translation is lost in a mouse model of Down's syndrome. J Neurosci [serie en Internet]. Oct 2010 [citado 5 Oct 2011]; 30(40): [aprox. 12 pantallas]. Disponible en: http://www.jneurosci.org/content/30/40/13537.full.pdf+html
7. Lyle R, Béna F, Gagos S, Gehrig C, Lopez G, Schinzel A, et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur J Hum Genet [serie en Internet]. 2009 [citado 5 Oct 2011]; 17(4): [aprox. 13 pantallas]. Disponible en: http://www.nature.com/ejhg/journal/v17/n4/pdf/ejhg2008214a.pdf
8. Netzer WJ, Powell C, Nong Y, Blundell J, Wong L, Duff K, et al. Lowering beta-amyloid levels rescues learning and memory in a Down syndrome model. PLoS One [serie en Internet]. Jun 2010 [citado 5 Oct 2011]; 5(6): [aprox. 5 pantallas]. Disponible en: http://www.plosone.org/article/fetchObjectAttachment.action;jsessionid=EEFEE4D2E7B6400883E6B61884332B11?uri=info%3Adoi%2F10
.1371%2Fjournal.pone.0010943&representation=PDF
9. Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses apc(min)-mediated tumours in mouse models of Down's syndrome. Nature [serie en Internet]. Ene 2008 [citado 5 Oct 2011]; 451(3): [aprox. 4 pantallas]. Disponible en: http://www.nature.com/nature/journal/v451/n7174/pdf/nature06446.pdf
10. Korbel JO, Tirosh-Wagner T, Eckehart Urban A, Chen XN, Kasowski M, Dai L, et al. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci [serie en Internet]. Jul 2009 [citado 5 Oct 2011]; 106(29): [aprox. 6 pantallas]. Disponible en: http://www.pnas.org/content/106/29/12031.full.pdf+html
11. Stankiewicz MJ, Crispino JD. ETS2 and ERG promote megakaryopoiesis and synergize with alterations in GATA-1 to immortalize hematopoietic progenitor cells. Blood [serie en Internet]. Abr 2009 [citado 5 Oct 2011]; 113(14): [aprox. 11 pantallas]. Disponible en: http://bloodjournal.hematologylibrary.org/content/113/14/3337.full.pdf+html
12. Jijón M. Genética y Síndrome de Down. Características generales. En: Síndrome de Down. Pautas mínimas para su entendimiento y atención. 2a ed. Quito: G&R Imprenta; 2010. p. 33-46
13. Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, et al. Synapjanine 1-linked phosphoinositide dishomeostasis and cognitive deficits in mouse models of Down's syndrome. Proc Natl Acad Sci [serie en Internet]. Jun 2008 [citado 5 Oct 2011]; 105(27): [aprox. 6 pantallas]. Disponible en: http://www.pnas.org/content/105/27/9415.full.pdf+html
14. Jijón M. El niño y niñas Down ante las enfermedades. Características generales. En: Síndrome de Down. Pautas mínimas para su entendimiento y atención. 2a ed. Quito: G&R Imprenta; 2010. p. 132-5
15. Kimura R, Kamino K, Yamamoto M, Nuripa A, Kida T, Kazui H, et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between â-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet [serie en Internet]. 2007 [citado 5 Oct 2011]; 16(1): [aprox. 9 pantallas]. Disponible en: http://hmg.oxfordjournals.org/content/16/1/15.full.pdf+html
16. Trazzi S, Mitrugno VM, Valli E, Fuchs C, Rizzi S, Guidi S, et al. APP-dependent up regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum Mol Genet [serie en Internet]. Ene 2011 [citado 5 Oct 2011]; 20(8): [aprox. 14 pantallas]. Disponible en: http://hmg.oxfordjournals.org/content/20/8/1560.full.pdf+html
Recibido: 13 de enero del 2013.
Aprobado: 23 de abril del 2013.
Dra. Odilkys Cala Hernández. Licenciada en Defectología. Master en Atención Integral al niño. Centro Provincial de Genética Médica. Pinar del Río. Correo electrónico: odigen@princesa.pri.sld.cu

{kind=link}
{kind=link}
{kind=link}
{kind=link}