










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
On-line version ISSN 1561-3194
Rev Ciencias Médicas vol.17 no.4 Pinar del Río July-Aug. 2013
PRESENTACIÓN DE CASO
Síndrome de distrofia miotónica tipo I
Myotonic dystrophy Type I
Pedro Ramírez Sosa, Vilma Esther Valladares Jiménez
Especialista de Primer Grado en Medicina General Integral. Máster en Asesoramiento Genético. Instructor. Policlínico "Ernesto Guevara de la Serna". Sandino. Correo electrónico: pedgenet@princesa.pri.sld.cu
RESUMEN
Introducción: la distrofia miotónica de Steinert, es una enfermedad multisistémica, autosómica dominante de penetrancia variable, causada por la expansión del triplete CTG, en el gen que codifica para la proteína kinasa de la distrofia miotónica en el cromosoma 19ql3. Se caracteriza por un fenómeno de anticipación, producto del cual su expresión es mayor en generaciones sucesivas y correlaciona con la talla de la expansión. Desde el punto de vista clínico se manifiesta por desórdenes multisistémicos asociados a disfunción muscular, siendo sus características más frecuentes la debilidad muscular lenta pero progresiva, atrofia muscular y el fenómeno miotónico.
Presentación del caso: paciente de 52 años de edad que fue visitado en su hogar durante el estudio de personas con discapacidad realizado en el cantón Quito, ya que tenía el diagnóstico de Distrofia Miotónica de Steinert a partir del cual se examina al resto de la familia y se encuentran signos y síntomas de la enfermedad en 3 familiares del propósito.
Conclusiones: no existe un sistema de asesoramiento genético en la comunidad que permita a la familia y al paciente con Distrofia Miotónica de Steinert la realización tanto de un seguimiento de su enfermedad como el conocimiento del nivel de recurrencia para este tipo de trastorno.
DeCS: Hipotonía muscular, Distrofia Miotónica tipo I, Enfermedad de Steinert.
ABSTRACT
Introduction: Steinert's Myotonic Dystrophy is a multiple-system, autosomal dominant of variable penetrance disease caused by an expansion of the cytosine-thymine-guanine (CTG) triplet, in the gene which codes for myotonic dystrophy protein kinase in chromosome19q13. It is characterized by a phenomenon known as anticipation, thus its expression is greater in successive generations and it correlates with the size of expansion. From the clinical view point it is expressed by multiple-system disorders associated with a muscle-dysfunction, the most frequent characteristics are muscle weakness it progresses slowly, muscle atrophy and myotonic phenomenon.
Case report: a 52-year old patient who was visited in his home during the study that involved disabled people in Canton, Quito, the patient suffered from Steinert's Myotonic Dystrophy,the the rest members of the family were as well examined; signs and symptoms of the disease were found in three other family members.
Conclusions: the study demonstrated the non-existence of a system to perform genetic counselling in the community, which would permit the follow-up of the families and patients suffering from Steinert's Myotonic Dystrophy, this difficulty also impedes the acquisition of knowledge regarding the level of recurrence for this type of genetic disorder.
DeCS: Muscular hypotonia, Myotonic dystrophy type I, Steinert disease.
INTRODUCCIÓN
La distrofia miotónica o enfermedad de Steinert es la distrofia muscular más frecuente en el adulto, con una prevalencia que se estima en 1/20000 habitantes y fue descrita en 1909 por Steinert, quien la consideró como una variante de la miotonía congénita, y en ese mismo año Ballén y Bibb la reconocieron como una entidad clínica única.1,2
Es una enfermedad heredo familiar que se transmite con patrón autosómico dominante y que tiene una expresividad clínica muy variable, observándose el fenómeno denominado anticipación y progresión de los síntomas, según el cual, en las sucesivas generaciones la enfermedad se manifiesta con mayor intensidad en etapas más tempranas que en las precedentes generaciones.2-4
A pesar de la variabilidad clínica de la distrofia miotónica tipo I, el gen defectuoso siempre ha sido el mismo en las poblaciones estudiadas. El daño tiene su locus afectado en el cromosoma 19q (19q13.2-13.3) y consiste en un defecto molecular; una secuencia de nucleótidos inestables (CTG) que es más larga en los pacientes afectados que en los no afectados. Mientras que los individuos normales tienen de 5 a 30 repeticiones de CTG, los pacientes con distrofia miotónica tienen de 50 a 2000. Los individuos con expansiones más grandes tienen más precoz el desorden y un fenotipo más severo.1,2,5,6
Es una enfermedad muscular de evolución lenta y progresiva que causa una afectación multisistémica que se manifiesta fundamentalmente en el músculo esquelético, siendo las características clínicas más relevantes la miotonía o dificultad para la relajación muscular después de una contracción muscular voluntaria o involuntaria, la atrofia o debilidad muscular lenta pero progresiva y las manifestaciones sistémicas.7,8
La debilidad muscular es la causante de la invalidez por degeneración de fibras musculares y se da fundamentalmente en los músculos faciales que le confiere a los individuos afectados una expresión característica (sonrisa invertida, mejillas hundidas y ptosis palpebral) y a nivel distal de miembros inferiores.8
Las manifestaciones sistémicas están dadas fundamentalmente por alteraciones cardiovasculares, gastrointestinales, respiratorias, endocrinometabólicas y casi todos los casos desarrollan cataratas5,7,8. Se describen cuatro formas clínicas de la enfermedad: leve, del adulto, de inicio en la infancia y distrofia miotónica tipo I congénita.9
Durante el estudio de personas con discapacidad realizado en el cantón Quito de la provincia de Pichincha, en Ecuador, encontramos una familia en la cual se presentan 4 casos de esta enfermedad con las características descritas en la literatura, en ella nos apoyamos para la realización de este trabajo.
PRESENTACIÓN DEL CASO
Durante el estudio de personas con discapacidad realizado en el cantón Quito de la provincia de Pichincha, en Ecuador, encontramos una familia en la cual se presentan 4 casos de Distrofia muscular tipo I o enfermedad de Steinert que presentaban las características descritas en la literatura.
A través del árbol genealógico se puede ver la herencia autosómica dominante que es característica de esta enfermedad. (Figura 1)
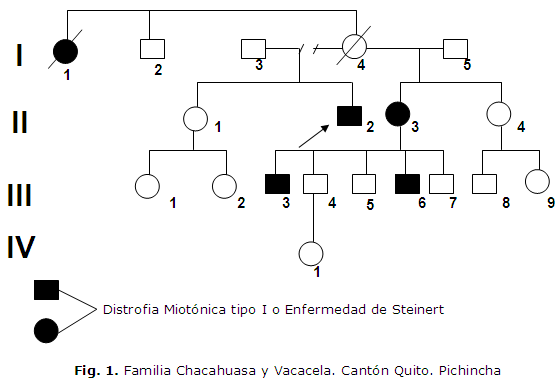
Al visitar la familia encontramos a un individuo de 52 años de edad que tenía el diagnóstico de Distrofia Miotónica Tipo I (II-2) y que presenta manifestaciones severas de la enfermedad, por lo que decidimos hacer un examen médico al resto de los integrantes de la familia previo consentimiento informado de cada uno de ellos, a través del cual se logró detectar signos y síntomas de la enfermedad en tres individuos (II-3,III-3 y III-6) que no conocían nada acerca de la misma, durante el interrogatorio se pudo constatar que la mujer (I-1) presentaba síntomas de la enfermedad antes de fallecer pero de la mujer (I-4) que es la madre del propósito y que también había fallecido no se pudo obtener datos a través del interrogatorio a la familia.
En esta familia los principales síntomas encontrados fueron:
Debilidad muscular fundamentalmente en los músculos faciales y a nivel distal en las extremidades tanto superiores como inferiores:
1. Dificultad para la relajación después de una contracción muscular provocada.
2. Paso de marcha.
3. Ptosis palpebral.
4. Sonrisa invertida.
Alteraciones Oftalmológicas
1. Cataratas.
Calvicie Precoz.
Manifestaciones Cardiovasculares (taquicardia).
Trastornos del sueño (somnolencia).
Mareos.
La edad de inicio de esta enfermedad en el caso de los individuos II-2 y II-3 comenzaron con la sintomatología aproximadamente a los 41 años de edad, pero los hijos de II-3 presentan una edad de inicio inferior observando que III-6, que tiene 16 años, ya presenta las manifestaciones de la enfermedad, lo que sugiere el fenómeno de anticipación genética presente en esta familia similar a lo reportado en la literatura revizada.4,7
Es de destacar que el individuo II-2 con un tiempo de evolución de 19 años tiene manifestaciones graves de la enfermedad, se encuentra en silla de ruedas y presenta complicaciones cardiovasculares, oftalmológicas y respiratorias severas que lo hacen totalmente dependiente del cuidado de su esposa, lo que demuestra que a medida que avance el tiempo de evolución las manifestaciones clínicas se hacen más severas y generalmente llevan al paciente a la invalidez total.7,8,10
En el momento que se realizó la visita se pudo constatar que el individuo II-2 ya había sido diagnosticado como enfermedad de Steinert, sin embargo no se habían realizado estudios y no se había orientado al resto de la familia sobre el riesgo de recurrencia de esta enfermedad ni el tratamiento oportuno por lo que existe falta de conocimiento sobre las causas, diagnóstico, tratamiento y evolución de la enfermedad por los familiares.
Las condiciones de vida del paciente y sus familiares son muy desfavorables agravando la calidad de vida de los pacientes con esta enfermedad, lo cual se informó en el puesto de mando para la posterior asistencia social por parte del gobierno a esta familia.
Por lo todo anterior se ofreció un buen asesoramiento genético a esta familia, basado en sus principios básicos, donde se informó todo lo relacionado con el diagnóstico, sintomatología, modo de herencia, evolución y tratamiento de la enfermedad, que a pesar del poco tiempo con que contamos para ello, se logró que la familia conociera los aspectos básicos para el cuidado y seguimiento de estos pacientes en el hogar así como las posibilidades de tratamiento en estos momentos los cuales se enumeran a continuación.
El tratamiento se concentra en manejar los síntomas y minimizar la discapacidad en la medida de lo posible.6,11,12
· Los bastones, las ortosis, los andadores y las sillas de ruedas motorizadas pueden ser útiles en los problemas de movilidad.
· Supervisión meticulosa de las funciones cardiaca y respiratoria que nos puede llevar a un tratamiento oportuno de estos problemas con un marcapasos cardíaco o un respirador portátil.
· Medicamentos y otros tratamientos para el estreñimiento y otros malestares del tracto digestivo.
· La cirugía de cataratas y la cirugía para los párpados caídos pueden mejorar la vista notablemente.
· Nuevos medicamentos para tratar la excesiva somnolencia pueden hacer la vida más fácil para la persona con DMM y su familia.
Ejercicios para el fortalecimiento del tono muscular.
Masajes tonificadores.
Utilización de diferentes texturas combinando las lisas con las rugosas, lo caliente con lo frío, etc.
Ejercicios para incrementar la percepción auditiva haciendo énfasis en la percepción fonemática y el oído fonemático, comenzando con los sonios del medio, para lograr adiestramiento del oído11.
Psicoterapia basada en el reconocimiento y aceptación de su enfermedad y las orientaciones para su enfrentamiento.10,11
Dinámica de familia.9-11
Interconsulta con otros especialistas para definir tratamiento medicamentoso según los síntomas que se van presentando.
Se concluye que no existe un sistema de asesoramiento genético que permita a la familia y al paciente con distrofia miotónica de Steinert la realización tanto de un seguimiento de su enfermedad como el conocimiento del nivel de recurrencia para este tipo de trastorno. Falta de un equipo multidisciplinario que evalúe a las personas con este tipo de distrofia miotónica y adecue un tratamiento, basándose en la sintomatología del paciente y teniendo en cuenta sus limitaciones, con vistas a atenuar sus síntomas y a elevar su calidad de vida.
Por lo que se recomienda la creación de un equipo multidisciplinario que valore al paciente y la familia con distrofia miotónica de Steinert, que permita el diagnóstico más tempranamente, así como su tratamiento oportuno y eficaz. A su vez incrementar los servicios de rehabilitación que permitan el acceso a los mismos por estos pacientes para con ello elevar su calidad de vida.
REFERENCIAS BIBLIOGRÁFICAS
1. Zaki M, Boyd PA, Impey L, Roberts A, Chamberlain P. Congenital myotonic dystrophy: prenatal ultrasound findings and pregnancy outcome. Ultrasound Obstet Gynecol[internet]. 2007[citado30 de noviembre 2011];29:284-8. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/uog.3859/full
2. De León MB, Cisneros B. Myotonic dystrophy 1 in the nervous system: from the clinic to molecular mechanisms. J Neurosci Res[internet] 2008 [citado 30 de noviembre 2011]; 86: 18-26. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/jnr.21377/full
3. Muraoka H, Negoro N, Terasaki F et al. Re-entry circuit in ventricular tachycardia due to focal fatty-fibrosis in a patient with myotonic dystrophy. Intern Med[internet]. 2005[citado 24 de noviembre 2011]; 44:129-135. Disponible en: http://ci.nii.ac.jp/search?q=Re-entry+circuit+in+ventricular+tachycardia+due+to++++focal+fatty-fibrosis+in+a+patient+with+myotonic+dystrophy&range=0&count=&sortorder=&type=0
4. Huson S. Myotonic dystrophy-time to improve patient care and prepare for pathogenesis based treatments. European Journal of Human Genetics[internet]. 2005[citado 30 de noviembre 2011]; 13(12):1312-1312. Disponible en: http://www.nature.com/ejhg/journal/v13/n12/full/5201500a.html
5. Martorell L, Cobo AM, Baiget M, Naud M, Poza, Parra J. Prenatal diagnosis in myotonic dystrophy type 1. Thirteen years of experience: implications for reproductive counseling in DM1 families. Prenatal Diagnosis[internet]. 2007[citado 21 de diciembre 2010]; 27: 68-72. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/pd.1627/abstract
6. Klein A, Clement E, Mercuri E, Muntoni F. Differential diagnosis of congenital muscular dystrophies. Eur J Paediatr Neurol[internet]. 2008[citado 30 de noviembre 2011]; 12: 371-7 Disponible en: http://www.sciencedirect.com/science/article/pii/S1090379807001869
7. Guimarães AS, Suazo GI, Nagahashi Marie SK. Fenómeno miotónico orofacial en pacientes con distrofia miotónica de Steinert. Avances en Odontoestomatología[internet]. 2010 May-Jun[citado 21 de septiembre 2011]; 26(3):139-142 Disponible en: http://europa.sim.ucm.es/compludoc/AA?articuloId=751369&donde=castellano&zfr=0
8. Schara U, Schoser BG. Myotonic dystrophies type 1 and 2: a summary on current aspects. Semin Pediatr Neurol[internet]. 2006[citado 22 de diciembre 2011]; 13: 71-9. Disponible en: http://www.sciencedirect.com/science/article/pii/S1071909106000921
9. Castel AL, Cleary JD, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol, 2010[citado diciembre 2010]; 11, 165-170. Disponible en: http://www.nature.com/nrm/journal/v11/n3/abs/nrm2854.html
10. Sayed AT, Moran PA. Myotonic dystrophy in pregnancy `a salutary tale'. J Obstet Gynaecol 2006[citado diciembre 2010]; 26(3): 258-60. Disponible en: http://cat.inist.fr/?aModele=afficheN&cpsidt=17694284
11. Argov Z, de Visser M. What we do not know about pregnancy in hereditary neuromuscular disorders. Neuromuscular Disorders 2009; 19(10): 675-9. Disponible en: http://www.sciencedirect.com/science/article/pii/S0960896609005719.
Recibido: 7 de noviembre del 2012.
Aprobado: 8 de mayo del 2013.
Dr. Pedro Ramírez Sosa. Especialista de Primer Grado en Medicina General Integral. Máster en Asesoramiento Genético. Instructor. Policlínico "Ernesto Guevara de la Serna". Sandino. Correo electrónico: pedgenet@princesa.pri.sld.cu
