










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
On-line version ISSN 1561-3194
Rev Ciencias Médicas vol.18 no.1 Pinar del Río Jan.-Feb. 2014
ARTICULO ORIGINAL
Programa de detección de errores innatos del metabolismo, Minas de Matahambre 2008-2012
Program of detection of innate errors of metabolism, Minas de Matahambre 2008-2012
Yinet Oliva López1, Raúl González García2
1Licenciada en Enfermería. Master en Asesoramiento Genético. Centro Municipal para el Desarrollo de la Genética Comunitaria. Minas de Matahambre. Correo electrónico: yinet@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral. Centro Municipal para el Desarrollo de la Genética Comunitaria. Minas de Matahambre. Profesor Instructor. Master en Asesoramiento Genético. Correo electrónico: raulgg@princesa.pri.sld.cu
RESUMEN
Introducción: los errores innatos del metabolismo son un grupo muy heterogéneo de enfermedades congénitas, determinadas por el bloqueo de un paso metabólico debido a la mutación de genes responsables del funcionamiento del mismo. Con herencia autosómico recesiva y en algunos casos ligada al cromosoma X, su diagnóstico precoz y el uso correcto de todas las opciones terapéuticas posibles es fundamental para asegurar la supervivencia y la mejor calidad de vida posible de los individuos afectados.
Objetivo: describir los resultados del programa de detección de errores innatos del metabolismo durante cinco años en el municipio Minas de Matahambre.
Material y Métodos: se realizó un estudio descriptivo y retrospectivo en el municipio de Minas de Matahambre del 2008 al 2012, al total de recién nacidos estudiados mediante el tamizaje neonatal.
Resultados: se estudiaron un total de 1822 recién nacidos, alcanzando una cobertura del 99,3 %. Existe mayor número de primeras determinaciones de: galactosa y 17 hidroxiprogesterona, no se han detectado casos confirmados de enfermos de hiperplasia adrenal congénita, fenilcetonuria, galactosemia, déficit de biotinidasa e hipotiroidismo congénito.
Conclusiones: con el presente trabajose describen los resultados del programa de detecci ón de errores innatos del metabolismo en el municipio, lo que permite el diagnóstico preciso, el correcto tratamiento médico y un adecuado asesoramiento genético a la familia.
DeCS: Errores innatos del metabolismo/epidemiología, Servicios de salud del niño.
ABSTRACT
Introduction: innate errors of metabolism are a very heterogeneous group of congenital diseases, determined by the blocking a metabolic passing due to mutation of genes responsible for the operation. Autosomal recessive inheritance and in some cases X-linked, early diagnosis and proper use of all possible treatment options is critical to assure the survival and the best possible quality of life of affected individuals.
Objective: to describe the results of the screening program of innate errors of metabolism for five years in the town of Minas de Matahambre.
Material and Methods: a retrospective descriptive study was conducted in the town of Minas de Matahambre from 2008 to 2012; the total number of infants were studied by neonatal screening.
Results: a total of 1822 infants were studied, reaching coverage of 99.3%. There is a greater number of first determinations: galactose and 17 hydroxyprogesterone were not detected in patients with confirmed congenital adrenal hyperplasia, phenylketonuria, galactosemia, biotinidase deficiency and congenital hypothyroidism cases.
Conclusions: the present work program results in detection of innate errors of metabolism in the town as being described, allowing accurate diagnosis, proper medical treatment and appropriate genetic counseling for the family.
DeCS: Metabolism inborn errors/epidemiology, Child health services.
INTRODUCCIÓN
Los errores innatos del metabolismo (EIM) o enfermedades metabólicas hereditarias, son un grupo muy heterogéneo de enfermedades congénitas,1,2 comprenden más de 200 afecciones monogénicas producidas por la deficiencia de una enzima funcional, un transportador de membrana o una proteína o cofactor específico.1,3 El bloqueo de la ruta metabólica provoca la acumulación de los sustratos sin degradar o la deficiencia de los productos finales, anomalías que originan distintos mecanismos fisiopatológicos. La causa del bloqueo es la mutación de genes responsables del funcionamiento de dicho paso metabólico, ya sea porque el producto génico, que puede ser una enzima o una coenzima, esté cualitativa o cuantitativamente afectado.1,2,3
Los EIM de los aminoácidos, los ácidos grasos y los ácidos orgánicos se manifiestan en los primeros años de vida mediante signos clínicos comunes, tales como letargia, falta de apetito, vómitos, taquipnea, convulsiones, trastornos del neurodesarrollo, entre otros, y pueden evolucionar hacia un cuadro clínico caracterizado por daño multisistémico grave, estupor, coma y un desenlace generalmente mortal.4
El mecanismo de herencia es, en la gran mayoría de los casos, autosómico recesivo y en contadas situaciones ligado al cromosoma X.3,5 Los ECM son afecciones genéticas individualmente raras pero colectivamente numerosas. Este número está aumentando constantemente en la medida en que se dispone de nuevos conceptos y técnicas para la identificación de fenotipos bioquímicos. Aunque son relativamente raros en la población pediátrica, estos trastornos han adquirido importancia creciente debido a que conducen a una elevada morbi-mortalidad y discapacidad.6
Se han descrito cerca de 500 EIM, y casi 25% de ellos afecta a los niños desde el periodo neonatal.2,3,4 Los EIM de manera individual, se han clasificado como enfermedades raras, siguiendo la definición internacionalmente aceptada de una frecuencia menor a 1 en 2,000, la incidencia conjunta de los EIM es significativa en la población infantil, 1:2 000 a 1:5 000,1:2 000, La mayor parte de los datos consistentes sobre la frecuencia de los EIM provienen de la información generada por los sistemas de tamiz neonatal ampliado de los países desarrollados.7 En cuanto a la incidencia, si bien cada vez hay mayor número de casos reportados, se considera que hay una subestimación de la misma dado que serían frecuentes los casos confundidos con otras afecciones más frecuentes o no detectados.1,3,6
Su diagnóstico depende primariamente de un alto índice de sospecha por parte del médico. Con este objetivo, se hace necesaria la aplicación de un método de evaluación clínica sencillo y de un protocolo de recolección de muestras. El primero permitirá plantear grupos de ECM para cada cuadro clínico, y las muestras biológicas, adecuadamente recolectadas y almacenadas, permitirán establecer un diagnóstico preciso.8
En 1983 se inició la detección neonatal de fenilcetonuria utilizando sangre seca en papel de filtro para medir la concentración de fenilalanina, por el método de Guthrie-Susi. En 1986 se extendió a todo el país el programa de pesquisa neonatal, y en el 2000 se introdujo una nueva tecnología en el pesquisaje a partir del uso del estuche diagnóstico UMTEST-PKU de producción cubana, a través del Sistema Ultra Micro Analítico (SUMA).9 En 1987 se dio inicio al pesquisaje de hipotiroidismo congénito a partir de sangre del cordón umbilical en el momento del nacimiento, en el 2006 se añadió a la pesquisa neonatal de fenilcetonuria a través del SUMA, el de tres nuevas enfermedades: la deficiencia de biotinidasa, galactosemia e hiperplasia adrenal congénita; en todos los casos, por tratarse de enfermedades genéticas, el diagnóstico preciso es indispensable para un adecuado asesoramiento genético a la familia. Para los padres del paciente siempre hay un alto riesgo de recurrencia de la misma situación.10
Para aquellos ECM con tratamiento específico, el diagnóstico es fundamental, en algunas formas de presentación aguda e intermitentes más tardías, el tratamiento de emergencia tiene por objeto disminuir la producción del metabolito tóxico proximal al punto de bloqueo, estimulando el anabolismo mediante aporte energético elevado. Una dieta que restrinja el producto que no puede metabolizarse es el tratamiento de elección para otras afecciones como la fenilcetonuria,11,12 hay un grupo de ECM que aún no tiene tratamiento específico.1,5,7
Teniendo en cuenta que la toma de la muestra se realiza al quinto día de nacido, obteniéndose los resultados en las tres primeras semanas de vida, lo que permite en caso positivo poner tratamiento oportuno a la enfermedad de que se trate y mejorar la calidad de vida de estos pacientes, con el objetivo de describir el comportamiento del programa de detección de errores congénitos del metabolismo mediante el tamizaje neonatal durante cinco años en el municipio de Minas de Matahambre, es que se realiza este trabajo.
MATERIAL Y MÉTODO
Se realizó un estudio longitudinal y retrospectivo en el municipio Minas de Matahambre, al total de recién nacidos durante el periodo de enero del 2008 a diciembre del 2012 a los cuales se les realizó el tamizaje neonatal, la sangre se obtuvo del talón del niño del quinto al décimo día de nacido y fue recogida sobre papel de filtro, se procesó a partir del uso del estuche diagnóstico UMTEST-PKU de producción cubana, a través del Sistema Ultra Micro Analítico (SUMA). La confirmación cuantitativa se realizó mediante la tecnología espectrofotométrica específica. Se determinaron los principales resultados del programa de detección de errores congénitos del metabolismo. Los resultados se presentan mediante tablas y gráficos.
RESULTADOS
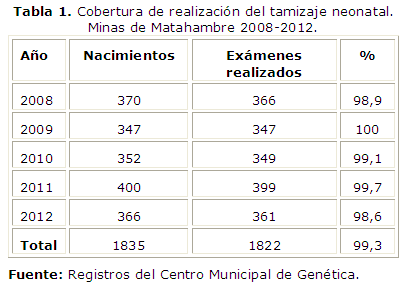
En la tabla 1 se muestra el total de estudios realizados durante el periodo analizado, la cobertura total fue del 99,3 %.
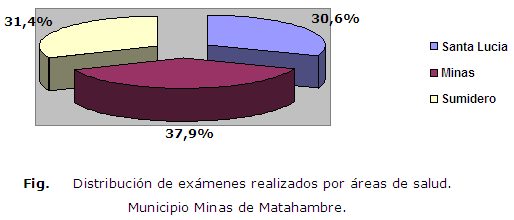
Teniendo en cuenta las tres áreas de salud existentes en el municipio, que son: Santa Lucia, Minas y Sumidero, la distribución de los exámenes realizados de forma decreciente fue de mayor proporción en el área de salud de Minas, Sumidero y Santa Lucia respectivamente (Figura).
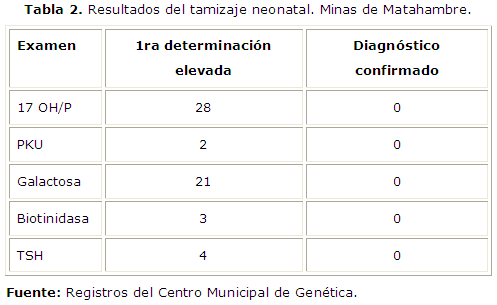
Durante el quinquenio analizado, existió un predominio de las primeras determinaciones elevadas de galactosa en sangre y de 17 hidroxiprogesterona, al realizarle el confirmatorio no se detectó ningún caso positivo para estas enfermedades como se muestra en la tabla 2 .
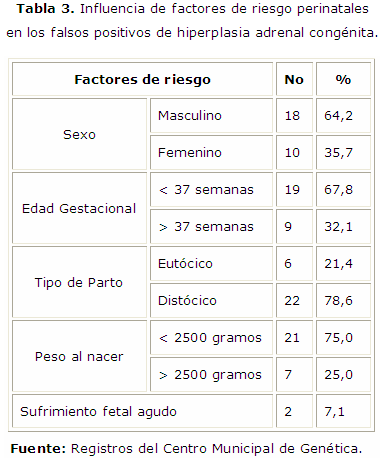
Se muestra en la tabla 3 que dentro de los factores de riesgo perinatales invocados en la elevación de los valores de 17OH/P y que constituyen falsos positivos predomina el sexo masculino, el parto pretérmino (antes de 37 semanas de gestación), la mayoría de ellos nacidos por cesárea, y en el 75,0 % el producto presentó un peso al nacer de menos de 1500 gramos, solo el 7,1 % presentó sufrimiento fetal agudo.
DISCUSIÓN
Durante el periodo 2008-2012 en Minas de Matahambre, la cobertura de realización del tamizaje neonatal a todos los recién nacidos fue del 99,3 %, influyendo en todos los casos el traslado después del nacimiento hacia otro lugar de residencia. En 1986 se extendió a todo el país el programa de pesquisa neonatal de fenilcetonuria por el método de Guthrie-Susi, y en el 2000 se introdujo una nueva tecnología en el pesquisaje a partir del uso del estuche diagnóstico UMTEST-PKU de producción cubana, a través del Sistema Ultra Micro Analítico (SUMA). En 1987 se dio inicio al pesquisaje de hipotiroidismo congénito a partir de sangre del cordón umbilical en el momento del nacimiento, a partir del 2005 la cobertura anual del programa de pesquisa neonatal de fenilcetonuria en el país supera el 97 %.9,10,13 En el 2006 se añadió el de tres nuevas enfermedades: la deficiencia de biotinidasa, galactosemia e hiperplasia adrenal congénita.10,13-16
La cobertura del pesquisaje de forma general para todas las enfermedades que incluye, alcanzada en el municipio, es superior a los reportes revisados, como en La Habana en el 2008 con el 95 %,14 en Granma 97 %.17 Teniendo en cuenta las áreas de salud, el área de Minas presentó una mayor cobertura del programa, coincide con el área de mayor número de nacimientos.
Durante el quinquenio estudiado existió un predominio de las primeras determinaciones elevadas para la hiperplasia adrenal congénita y para la galactosemia. Al realizarle el confirmatorio no se detectó ningún caso positivo para estas enfermedades. Es de interés señalar que durante el período analizado, en la hiperplasia adrenal congénita se estudiaron 1822 recién nacidos y en 28 de ellos se obtuvo elevación de la primera muestra, representando el 1,53 %, siendo menor que lo obtenido en Cuba durante enero del 2005 a junio del 2010, donde se estudiaron 548 838 recién nacidos, de los cuales 10 028 presentaron la primera determinación elevada con un 1,82 %.17 Se plantean tasas de falsos positivos por encima de 1 %,18 entre las posibles causas se encuentran: la prematuridad, el bajo peso al nacer, las enfermedades graves, dentro de ellas los trastornos respiratorios, la pobre función renal o hepática, el estrés quirúrgico, metabólico o traumático. También se plantean otras causas como la toma de la muestra con menos de 48 horas de vida, la baja especificidad de algunos antisueros utilizados en los inmunoensayos, así como las reacciones cruzadas con otros esteroides.13
En el municipio durante el periodo analizado se presentó un predominio de casos con valores elevados de 17OH/P que posteriormente se normalizaron, por lo que constituyen falsos positivos (28). Teniendo en cuenta que los costos económicos y psicosociales asociados a estos son elevados, se determinó la influencia de algunos factores de riesgo invocados, predominando el sexo masculino, el parto pretérmino (antes de las 37 semanas de gestación), la mayoría de ellos nacidos por cesárea y predominó el bajo peso al nacer (menos de 2500 gramos). Es significativo que solo un 7,1 % presentó sufrimiento fetal agudo, esto se corresponde con el estudio realizado desde el año 2007 al 2010 en La Habana. De forma general se plantea que existen diferencias según el sexo, probablemente los varones sanos tienen niveles más altos de 17OHP. Esto se ha propuesto como explicación de la preponderancia del sexo masculino.13
En relación con el tipo de parto, predominó el nacimiento por cesárea. Se señala que los nacidos por partos distócicos presentaron niveles de 17OHP, ligeramente superiores a los de parto eutócico. Esto pudiera estar en relación con los eventos perinatales desfavorables, que se asocian con frecuencia a las distocias del parto. También predominó en pretérminos y en el bajo peso al nacer, en ellos existe una disminución de la actividad de la 3-beta-hidroxiesteroide-deshidrogenasa, y cuando son menores de 30 semanas de edad gestacional aparece una disminución de la actividad de la 11-beta-deshidrogenasa o un retraso en la expresión de esta enzima, lo cual justifica la elevación de dicho metabolito. Se relaciona además con una degradación de 17OHP disminuida por inmadurez de la función hepática, a lo que se suma una producción aumentada de la hormona a causa del estrés al que están sometidos estos neonatos.13
El presente trabajo ha posibilitado describir el comportamiento del programa de detección precoz de errores innatos del metabolismo en el municipio Minas de Matahambre durante un quinquenio, lo que permite un diagnóstico oportuno y con ello un seguimiento adecuado de los casos positivos.
REFERENCIAS BIBLIOGRÁFICAS
1. Vela M, Belmont L, Fernández C, Ramírez C, Ibarra I. Frecuencia de enfermedades metabólicas congénitas susceptibles de ser identificadas por el tamiz neonatal. Acta Pediatr Mex [internet]. 2009 [citado 2 Mayo de 2013]; 30(3): [aprox. 7 p.]. Disponible en: http://www.medigraphic.com/pdfs/actpedmex/apm-2009/apm093e.pdf
2. Juan-Fita MJ, Egea JM, González I, Moya MR, Fernández A. Cribado neonatal ampliado en la Región de Murcia. Experiencia de tres años. Medicina Clinica [internet]. 2011 [citado 2 Mayo de 2013]; [aprox. 7 p.]. Disponible en: http://zl.elsevier.es/es/revista/medicina-clinica-2/pdf/90161780/S300/
3. Pintos G. Déficit de biotinidasa: las dos caras del cribado metabólico. Med Clin (Barc) [internet]. 2011 [citado 2 Mayo de 2013]; 20(10): [aprox. 3 p.]. Disponible en: http://www.elsevier.es/sites/default/files/elsevier/eop/S0025-7753(11)00660-9.pdf
4. Hanley WB. Non-PKU mild hyperphenylalaninemia (MHP)—The dilemma. Molecular Genetics and Metabolism, 2011; 104(1): 23-26.
5. Galbe J, Grupo Previnfad/Papps infancia y adolescencia. Cribado neonatal de metabolopatías. Rev Pediatr Aten Primaria [internet]. 2009 [citado 2 Mayo de 2013]; 11(43): [aprox. 14 p.]. Disponible en: http://scielo.isciii.es/pdf/pap/v11n43/9_previnfad.pdf
6. Loukas YL, Soumelas GS, Dotsikas Y, Georgiou V, Molou E, Thodi G, et al. Expanded newborn screening in Greece: 30 months of experience. Journal of inherited metabolic disease, 2010; 33 Suppl 3: S341-8. Available from: http://link.springer.com/article/10.1007%2Fs10545-010-9181-8
7. Alfonso I, Charria G, Papazian O. Estado actual de la pesquisa neurometabólica neonatal. Medicina (B. Aires) [internet]. 2009 [citado 2 Mayo de 2013]; 69(1): [aprox. 5 p.]. Disponible en: http://www.scielo.org.ar/pdf/medba/v69n1s1/v69n1s1a05.pdf
8. Agulló AG, Calvet EV, Lezcano AC, Bargadá M, Vilalta NP. Crecimiento y maduración de los pacientes con hipotiroidismo congénito detectados por el programa de cribado neonatal en Cataluña (1986-1997). Medicina clínica. 2010[citado 2 Mayo de 2013]; 134(7): 287-295. Disponible en: http://zl.elsevier.es/es/revista/medicina-clinica-2/crecimiento-maduracion-los-pacientes-hipotiroidismo-congenito-detectados-13148191-originales-2010
9. Perdomo JC, Luna E, Domínguez ME, Castro M, Rodríguez D, Landa M, Ravelo O, Monzón M. El programa de diagnóstico, manejo y prevención de enfermedades genéticas y defectos congénitos en la provincia de matanzas: 1988-2008. Rev Cubana Genet Comunit. [internet]. 2009 [citado 20 de Junio de 2012]; 3(2,3): [aprox. 8 p.]. Disponible en: http://www.bvs.sld.cu/revistas/rcgc/v3n2_3/rcgc0523010%20esp.html
10. Marcheco B. El Programa Nacional de Diagnóstico, Manejo y Prevención de Enfermedades Genéticas y Defectos Congénitos de Cuba: 1981-2009. Rev Cubana Genet Comunit [internet]. 2009 [citado 2 Mayo de 2013]; 3(2,3): [aprox. 18 p.]. Disponible en: http://bvs.sld.cu/revistas/rcgc/v3n2_3/cuba.pdf
11. Vela M, Ibarra I, Monroy S, Fernández C, Guillén S, Belmont L, et al. Modelo de atención inicial de la fenilcetonuria y otras hiperfenilalaninemias en el Instituto Nacional de Pediatría. Acta Pediatr Mex, [internet]. 2010 [citado 2 Mayo de 2013]; 31(6): [aprox. 7 p.]. Disponible en: http://www.medigraphic.com/pdfs/actpedmex/apm-2010/apm106h.pdf
12. Alcantara MA, García B, Barrientos R, González A. Aspectos generales y panorama actual del estudio molecular de la fenilcetonuria (PKU) en México. Acta Pediatr Mex [internet]. 2012 [citado 2 Mayo de 2013]; 33(6): [aprox. 4 p.]. Disponible en: http://www.medigraphic.com/pdfs/actpedmex/apm-2012/apm126m.pdf
13. Espinosa TM, Hernández M, Carvajal F, González E, Domínguez E. Influencia de factores perinatales en la pesquisa neonatal de hiperplasia adrenal congénita en Ciudad de La Habana y La Habana. Rev Cubana Endocrinol. [internet]. 2012 [citado 2 Mayo de 2013]; 23(1): [aprox. 8 p.]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1561-29532012000100001
14. Cedré MA, Herrera A, Barrios JC. Programa de diagnóstico, manejo y prevención de enfermedades genéticas y defectos congénitos en la provincia La Habana: 1988-2008. Rev Cubana Genet Comunit [internet]. 2009 [citado 2 Mayo de 2013]; 3(2,3): [aprox. 1 p.]. Disponible en: http://bvs.sld.cu/revistas/rcgc/v3n2_3/rcgc0323010%20esp.html
15. Dyce E, Moras F, Pimentel H. El programa de diagnóstico, manejo y prevención de enfermedades genéticas y defectos congénitos en la provincia Camagüey: 1984-2007. Rev Cubana Genet Comunit [internet]. 2009 [citado 2 Mayo de 2013]; 3(2,3): [aprox. 6 p.]. Disponible en: http://bvs.sld.cu/revistas/rcgc/v3n2_3/camaguey.pdf
16. Bay LB, De Pinho S, Eiroa HD, Otegui I, Rodríguez R. La importancia de una ley a tiempo: presentación de un caso de deficiencia de biotinidasa no diagnosticado por pesquisa neonatal. Arch. argent. pediatr. [internet]. 2010 [citado 2 Mayo de 2013]; 108(1): [aprox. 4 p.]. Disponible en: http://www.scielo.org.ar/pdf/aap/v108n1/v108n1a17.pdf
17. Martínez ML, Ravelo I, La O A, Hormigó L, Vargas I, Meriño G. El Programa de Diagnóstico, Manejo y Prevención de Enfermedades Genéticas y Defectos Congénitos en la Provincia Granma. Años 1986-2009. Memorias Convención Internacional de Salud Pública. Cuba Salud [internet]. 2012 [citado 2 Mayo de 2013]; [aprox. 6 p.]. Disponible en: http://www.convencionsalud2012.sld.cu/index.php/convencionsalud/2012/paper/view/1633
18. Zaldivar JR, Rodríguez A, Quesada M, Navarro A, Martínez M. Hiperplasia adrenal congénita clásica: Tratamiento médico y quirúrgico. MEDISAN [internet]. 2010 [citado 2 Mayo de 2013]; 14(6): [aprox. 5 p.]. Disponible en: http://scielo.sld.cu/pdf/san/v14n6/san17610.pdf
Recibido: 30 de agosto del 2013.
Aprobado: 22 de octubre del 2013.
Lic. Yinet Oliva López. Licenciada en Enfermería. Master en Asesoramiento Genético. Centro Municipal para el Desarrollo de la Genética Comunitaria. Minas de Matahambre. Correo electrónico: yinet@princesa.pri.sld.cu
