










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
On-line version ISSN 1561-3194
Rev Ciencias Médicas vol.18 no.1 Pinar del Río Jan.-Feb. 2014
PRESENTACIÓN DE CASO
Tumor intracardiaco en el recién nacido
Intracardial tumor in the newborn
Yanett Sarmiento Portal1, Omar León Vara Cuesta2, Yordis Mailin Gutiérrez Cruz3, Angelicia Crespo Campos4, Maria Elena Portal Miranda5
1Especialista de Primer Grado en Medicina General Integral. Especialista de segundo Grado en Neonatología. Investigadora agregada. Asistente. Máster en Atención Integral al Niño. Hospital General Docente Abel Santamaría Cuadrado. Pinar del Río. Correo electrónico: yanencesa@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral. Especialista de segundo Grado en Neonatología. Investigador agregado. Auxiliar. Máster en Atención Integral al Niño y en Urgencias Médicas. Hospital General Docente Abel Santamaría Cuadrado. Pinar del Río. Correo electrónico: belkys@princesa.pri.sld.cu
3Especialista Primer Grado en Medicina General Integral. Residente tercer año neonatología. Correo electrónico: mailin@has.sld.cu
4Especialista de Primer Grado en Medicina General Integral y Neonatología. Investigador agregado. Asistente. Máster en Atención Integral al Niño. Hospital General Docente Abel Santamaría Cuadrado. Pinar del Río. Correo electrónico: angelicia@princesa.pri.sld.cu
5Especialista de Segundo Grado en Neonatología. Asistente. Máster en Atención Integral al Niño. Hospital General Docente Abel Santamaría Cuadrado. Pinar del Río. Correo electrónico: portal23@princesa.pri.sld.cu
RESUMEN
Introducción: los tumores primarios cardiacos son raros durante la infancia y en su mayoría benignos, siendo los rabdomiomas los más comunes, asociándose en más del 60% de los casos con esclerosis tuberosa. La mayoría de ellos tienden a involucionar, pero algunos, según su localización y manifestaciones clínicas, requerirán tratamiento quirúrgico.
Caso clínico: recién nacido del sexo femenino, hija de madre de 34 años. Nace producto de cesárea iterada a las 40 semanas, Apgar 9-9 y peso al nacer 2800 gramos. Tuvo seguimiento por Genética Clínica durante el embarazo por detectarse en ultrasonido prenatal la presencia de tumoración intracardiaca, la cual se confirma al realizar ecografía postnatal, con un área tumoral de 3,5mm. Se diagnostica Rabdomioma intracardiaco que no obstruye el tracto de salida, con seguimiento clínico y ecocardiográfico mensual y evolución favorable.
Conclusiones: los tumores cardiacos fetales son extraordinariamente raros. Se pueden diagnosticar por ecografía desde la vida intrauterina. La actitud recomendada es expectante por la posibilidad de regresión espontánea, excepto en aquellos casos con repercusión clínica. En el seguimiento se debe descartar la presencia de esclerosis tuberosa por su elevada asociación con esta entidad.
DeCS: Neoplasias, Rabdomioma, Recién nacido, Diagnóstico prenatal, Esclerosis tuberosa.
ABSTRACT
Introduction: primary cardiac tumors are rare in childhood and mostly benign, the most common being rhabdomyomas, associating more than 60% of patients with tuberous sclerosis. Most of them tend to regress, but some, depending on their location and clinical manifestations, require surgical treatment.
Case presentation: newborn female, daughter of a mother of 34 years old. Iterated product cesarean birth at 40 weeks, Apgar 9-9, 2800 grams of birth weight. Clinical genetics was tracking during pregnancy by prenatal ultrasound detected in the presence of Intracardial tumor, which was confirmed by postnatal ultrasound performed with a tumor area of 3.5 mm. Intracardial rhabdomyomas diagnosed does not obstruct the outflow tract, with clinical and echocardiography follow-up and monthly favorable.
Conclusions: fetal cardiac tumors are extremely rare. They can be diagnosed by ultrasound from the womb. The recommended expectant attitude is the possibility of spontaneous regression, except in cases with clinical impact. At follow-up, the tuberous sclerosis must be ruled out by its high association with this entity.
DeCS: Neoplasms, Rhabdomyoma, Newborn infant, Prenatal diagnosis, Tuberous sclerosis.
INTRODUCCIÓN
Los tumores cardíacos primarios constituyen una patología muy poco frecuente a toda edad, pero por la condición del órgano que comprometen, tienen una especial relevancia clínica y un pronóstico poco predecible. 1
La incidencia de tumores cardíacos en la infancia varía entre 0,0017 y 0,28%. Más del 90% son de naturaleza benigna. La variedad más frecuente es el rabdomioma, el que se asocia en más del 60% de los casos con esclerosis tuberosa. Otros tumores son el fibroma (30-50%), la cardiomiopatía histiocitoide (<10%), teratoma (<10%), sarcoma (<10%) hemangioma (<5%) y el mixoma (<5%). 2-4
El primer rabdomioma fue descrito por Von Recklinghausen en 1862. Hasta los años setenta el estudio de elección para su diagnóstico era el angiocardiograma. Posteriormente éste fue sustituido por el ecocardiograma y en 1982 De Vore hizo el diagnóstico de tumor in útero. Desde entonces se han incrementado los reportes de estas lesiones detectadas desde la vida fetal. 5,6
El diagnóstico de tumores cardiacos durante la vida fetal es muy infrecuente, con una prevalencia aproximada de 1:10.000 nacidos vivos, lo que representa un 1% del total de las alteraciones cardiacas detectadas.6,7
El diagnóstico se realiza habitualmente entre las 20-30 semanas de gestación, mediante estudio ecográfico. La repercusión fetal está determinada por la localización y el tamaño de la tumoración, que puede comprometer el flujo sanguíneo, interferir con la función miocárdica o desencadenar arritmias cardiacas que pueden llevar a la muerte en la vida intrauterina o el desarrollo de hidrops. Se ha informado la terminación del embarazo hasta en un 57% de los casos. 3,5
Debido a que se trata de una entidad extremadamente rara, se presenta el caso de un paciente con rabdomioma intracardiaco cuyo diagnóstico se realizó en la etapa prenatal.
PRESENTACIÓN DEL CASO
Recién nacido del sexo femenino, hija de madre de 34 años de edad con antecedentes de migraña, G3 A1 C1. Nace producto de cesárea iterada a las 40 semanas de gestación, Apgar 9-9 y un peso al nacer de 2800 gramos. Tuvo seguimiento por Genética Clínica durante el embarazo por detectarse en ultrasonido prenatal la presencia de tumoración intracardiaca.
Nace en buenas condiciones y es trasladado a la Unidad de Cuidados Intensivos Neonatales para corroborar diagnóstico prenatal.
Se recibió rosado, bien perfundido, sin signos de distrés respiratorio y con respuesta neurológica adecuada. Se realizaron complementarios que incluyeron Grupo y factor, hematocrito, glicemia, ionograma, gasometría, telecardiograma y electrocardiograma los cuales resultaron normales. Se interconsultó el caso con Genética clínica no encontrándose rasgos dismórficos. Se realizó ultrasonido de cráneo y abdomen sin alteraciones.
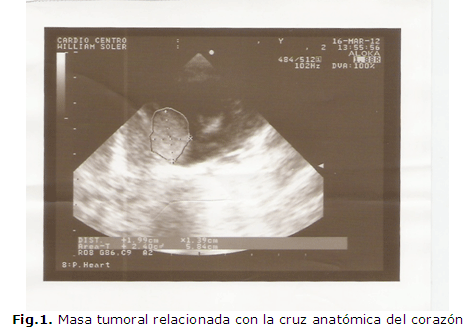
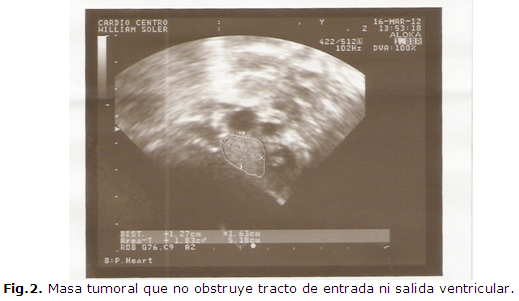
Se valoró además con cardiología pediátrica, realizándose ecocardiograma (Figura 1 y Figura 2) donde se comprueba masa tumoral relacionada con la cruz anatómica del corazón, con las siguientes dimensiones:
Área tumoral: 3.15 mm
Volumen tumoral: 4.1 mm
Peso tumoral: aproximadamente 4.3 gramos
No se comprueba obstrucción de los tractos de entrada ni salida ventriculares Comunicación interauricular tipo foramen oval de 1.6 mm.
Se realizó evaluación conjunta en el Cardiocentro "William Soler", decidiéndose mantener conducta expectante ante la ausencia de sintomatología clínica y la estabilidad hemodinámica, con seguimiento clínico y ecocardiográfico mensual.
Se egresa a los 15 días de vida con evolución favorable y con el diagnóstico de Rabdomioma aislado que no obstruye tracto de salida.
DISCUSIÓN
Los tumores cardiacos pueden ser diagnosticados durante la vida fetal en la segunda mitad de la gestación. De ellos, el más frecuente es el rabdomioma, aunque hasta el momento se han descrito en la bibliografía médica internacional menos de 300 casos. 6,8
Los rabdomiomas cardíacos por lo general son solitarios y silentes, con tendencia a la regresión, pero pueden llegar a producir complicaciones que van desde arritmias, obstrucción al flujo sanguíneo e incluso muerte súbita. La localización auricular es rara, siendo más frecuentes a nivel ventricular principalmente izquierdo, músculos papilares y en relación con el septo interventricular, como en el caso que se describe, sitios en los cuales, dependiendo de su tamaño, podrán obstruir tractos de entrada o salida que pueden comprometer el funcionamiento cardíaco. 4 La literatura describe relación entre el síndrome de Wolff Parkinson White y la localización auricular de los rabdomiomas, más frecuente en varones y durante el primer año de vida.9
El rabdomioma se caracteriza por ser circunscrito, lobulado, blanquecino o grisáceo. Su tamaño varía desde unos milímetros hasta algunos centímetros; en 10% de los casos aparece como una masa endocavitaria múltiple o única, y a veces se considera un hamartoma. Histológicamente es benigno, está formado por células aracniformes: células musculares con abundante glicógeno y de éste el sarcoplasma aparece a manera de prolongaciones desde el núcleo a la periferia.6
La mayoría de los rabdomiomas cardiacos aislados diagnosticados prenatalmente muestran una evolución favorable, con una regresión y una resolución completa sin complicaciones durante los primeros años de la infancia en el 80% de los casos, aunque suele asociarse con esclerosis tuberosa entre un 63 y un 80% según las series publicadas, por lo que se pueden considerar como un marcador ecocardiográfico prenatal de dicha enfermedad. Esta asociación aumenta en caso de historia familiar previa o multifocalidad de los tumores. 3,4,6
La esclerosis tuberosa, también conocida como enfermedad de Bouneville Pringle, es una enfermedad neurocutánea con patrón de herencia autosómico dominante, con alta penetrancia y variabilidad. Se caracteriza por la aparición de hamartomas en múltiples sistemas. Sus principales manifestaciones clínicas incluyen: angiofibromas faciales, máculas hipomelanóticas, nevus del tejido conectivo, lesiones en piel y faneras, túberes corticales, nódulos subependimales, astrocitomas de células gigantes, líneas de migración radial en la sustancia blanca cerebral, hamartoma nodular múltiple en los ojos, angiomiolipoma y quistes renales, agujeros en esmalte dental, fibromas gingivales y los rabdomiomas descritos. Las manifestaciones clínicas usualmente no aparecen desde el nacimiento y se van observando con el trascurrir de los años. 4,6,10
Por lo tanto, el seguimiento clínico de los pacientes con manifestaciones que sugieren esta condición, como el rabdomioma cardiaco en el caso que se presenta, es indispensable para tomar las conductas apropiadas en el momento indicado.
A pesar de que por mucho tiempo la angiografía fue el patrón de oro para realizar el diagnóstico de los tumores cardiacos, desde hace varios años el ultrasonido cardiaco, la tomografía computarizada y la resonancia magnética, se consideran como herramientas más útiles y han reemplazado a las antiguas técnicas invasivas. 4,10
En el diagnóstico diferencial de tumoración cardiaca hay que incluir otras entidades, como los teratomas, hemangiomas, fibromas y mixomas. Los teratomas tienen localización habitualmente extracardiaca (a nivel del pericardio y de la raíz de las arterias pulmonar y aorta). Suelen presentarse como tumoraciones únicas, heterogéneas y pediculadas. Pueden presentarse clínica mente por compresión (fracaso cardiaco, derrame pericardico, hydrops fetal no inmune y muerte fetal). Los fibromas se localizan a nivel del miocardio ventricular, suelen ser lesiones únicas, sólidas, isoecogénicas con el tejido miocárdico adyacente y pueden tener calcificaciones centrales. Presentan crecimiento en el periodo posnatal. Los hemangiomas son tumores intracardiacos poco frecuentes, que se localizan próximos a la aurícula derecha o a las venas. Se asocian a mesotelioma o rabdomiosarcoma. Clínicamente pueden asociarse a un derrame pericárdico y ecográficamente se presentan como lesiones sésiles de ecogenicidad mixta. Los mixomas cardiacos fetales son excepcionales, a diferencia de lo que ocurre en los tumores cardiacos primarios del adulto. Su comportamiento suele ser bifásico, con un crecimiento progresivo hasta la semana 32 de gestación, y posteriormente tiende a reducirse progresivamente durante los dos primeros años de vida. Los rabdomiomas pueden ser localizados o multifocales, suelen aparecer en el tabique interventricular o en la pared libre ventricular y auricular. Su apariencia ecográfica es la de una masa redondeada hiperecogénica, bien definida. 3,5,7,8 Estas características hicieron que el caso presentado fuera diagnosticado de probable rabdomioma cardiaco, aunque no se dispone de la confirmación histológica por no haberse realizado tratamiento quirúrgico.
Los indicadores de un mal pronóstico son el desarrollo de obstrucción al flujo, alteración de la válvula atrioventricular con regurgitación, presencia de arritmias cardiacas, insuficiencia cardiaca y desarrollo de hydrops fetal. Por ello, aunque la mayoría de estos tumores son benignos, su pronóstico no depende sólo de la naturaleza histológica, sino también de la localización, el tamaño, el número de tumores y las complicaciones asociadas.3
Debido a la historia natural de la regresión espontánea de los rabdomiomas, éstos deben tratarse de forma expectante. La terapia quirúrgica sólo se indica para los síntomas secundarios a una masa específica. La presencia de obstrucción a la salida ventricular, obstrucción a la afluencia de la válvula atrioventricular, o las arritmias intratables son indicaciones para resecar estos tumores. La meta de la terapia quirúrgica es resecar únicamente la masa o porción de masa responsable de la obstrucción y causante de la sintomatología adversa. Es importante no dañar las estructuras a las cuales está adherido el tumor. La resección completa no es necesaria. Típicamente, el tejido rabdomiomatoso restante regresará de manera espontánea y no causará ninguna otra secuela.2,3,4,6,8,10
CONCLUSIONES
- Los tumores cardiacos fetales son extraordinariamente raros. Se pueden diagnosticar por ecografía desde la vida intrauterina. La actitud recomendada es expectante por la posibilidad de regresión espontánea, excepto en aquellos casos con repercusión clínica. En el seguimiento se debe descartar la presencia de esclerosis tuberosa por su elevada asociación con esta entidad.
REFERENCIAS BIBLIOGRÁFICAS
1. Arnaiz GP, Toledo GI, Borzutzky SA, Urcelay MG, Heusser RF, Garay GF. Comportamiento clínico de los tumores cardíacos desde el feto hasta el adulto: serie multicéntrica de 38 pacientes. Rev. méd. Chile [revista en la Internet]. 2006 Sep [citado 2013 Ago 15]; 134(9): 1135-1145. Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0034-98872006000900008&lng=es.
2. Rios J. Mixoma cardíaco con diagnóstico prenatal: Presentación de un caso y revisión de literatura. Rev Med Hered [online] 2012;23(4): 247-250. [citado 2013-07-26]. Disponible en: http://www.scielo.org.pe/scielo.php?pid=S1018-130X2012000400007&script=sci_arttext
3. López N, Rodríguez R, Vegas G, De La Calle M, González A. Tumores cardiacos fetales: diagnóstico ecográfico, evolución y tratamiento. Rev chil obstet ginecol 2011; 76(3): 147-154. [citado 2013-07-26] Disponible en: http://www.scielo.cl/pdf/rchog/v76n3/art03.pdf
4. Arango Posada CA. Rabdomiomas cardíacos y Esclerosis tuberosa: Presentación de dos casos en recién nacidos. Arch Med (Manizales) 2012; 12(2): 199-204. [citado 2013 Ago 15]. Disponible en: http://revistasum.umanizales.edu.co/ojs/index.php/archivosmedicina/article/viewFile/11/11
5. Miranda Chávez I, Muñoz Castellanos L, Buendía Hernandez A, Aranda Faustro A, Erdmenger Orellana J, Ramírez Marroquín Sl. Rabdomioma gigante intracardíaco en la etapa neonatal. Reporte de un caso. Arch. Cardiol. Méx. [revista en la Internet]. 2004 Mar [citado 2013 Oct 22]; 74(1): 49-52. Disponible en: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S1405-99402004000100007&lng=es.
6. Anaya Reyes P, Rodríguez Rábago MJ. Diagnóstico prenatal de rabdomioma cardiaco. Reporte de un caso. Ginecol Obstet Mex 2013;81:477-481. Sitio en internet Google. [citado 2013-07-26] Disponible en: http://www.medigraphic.com/pdfs/ginobsmex/gom-2013/gom138h.pdf
7. Savío Benavides A, Oliva Rodríguez J, García Morejón C, García Guevara C, Arencibia Faire J, Ponce Bittar J. Diagnóstico ecocardiográfico de los tumores primarios del corazón en el feto. Rev Cubana Pediatr [revista en la Internet]. 2009 [citado 2013 Jul 26]; 81(4):1-9. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312009000400001&lng=es.
8. Chao AS, Chao A, Wang TH, Chang YC, Chang YL, Hsieh CC, et al. Outcome of antenatally diagnosed cardiac rhabdomyoma: Case series and a meta-analysis. Ultrasound Obstet Gynecol. 2008;31:289-95. [citado 2013 Oct 22] Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/uog.5264/full
9. Tejero Hernández MA, Gómez Guzmán E, Tejero Mateos I, Pérez Navero JL, Suarez de Lezo Cruz Conde J. Rabdomioma auricular derecho y syndrome de Wolff-Parkinson-White en una lactante con esclerosis. J an ped. 2009; 1(1):500-2. [citado 2013 Oct 22] Disponible en: http://zl.elsevier.es/es/revista/anales-pediatria-37/articulo/rabdomioma-auricular-derecho-sindrome-wolff-parkinson-white-13137050
10. Lince RL, Gómez López de Mesa C, Arteaga A, Montoya JH, Vásquez LM. Rabdomioma cardiaco como manifestación de esclerosis tuberosa. Presentación de dos casos y revisión de la literatura. Rev. Colom. Cardiol.[serial on the Internet]. 2009 Oct [cited 2013 Aug 15];16(5): 224-228. Available from: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0120-56332009000500006&lng=en.
Recibido: 9 de septiembre del 2013.
Aprobado: 28 de noviembre del 2013.
Dra. Yanett Sarmiento Portal. Especialista de Primer Grado en Medicina General Integral. Especialista de segundo Grado en Neonatología. Investigadora agregada. Asistente. Máster en Atención Integral al Niño. Correo electrónico: yanencesa@princesa.pri.sld.cu
