










 Custom services
Custom services text in
text in  Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Los microsatélites son secuencias cortas repetidas en tándem (conocidas por sus acrónimos STR de short tandem repeats o SSR de simple sequence repeat, por sus siglas en inglés) con unidades de repetición entre 1 y 6 pb (algunos autores extienden su definición hasta 8 pb), que pueden constituir tractos de repeticiones que van desde algunas copias hasta cientos de éstas. Estas secuencias son abundantes en los genomas de eucariotas, asociadas fundamentalmente a regiones no codónicas, aunque no privativas de éstas. También se encuentran presentes en genomas procariotas constituyendo importantes marcadores para la genotipificación, clasificación y control epidemiológico de especies de interés 1. Entre las principales motivaciones para el estudio de estas secuencias se encuentran su participación en procesos tales como la recombinación y la regulación de la transcripción 2, y cuando se encuentran en regiones codificantes son causa de afecciones neurodegenerativas como síndrome de frágil X, enfermedad de Huntington (HD), atrofia espinobulbar-muscular (SBMA), el síndrome de Haw River (DRPLA), las ataxias espinocerebelosas (SCA1, SCA2, SCA3, SCA6, SCA7 y SCA17), así como algunos tipos de cáncer 3,4. En el desarrollo de vacunas se ha demostrado cómo gérmenes patógenos pueden evadir la respuesta inmune simplemente alterando la composición de las secuencias repetidas en sus genes 5. Se ha demostrado cómo la expansión y contracción de microsatélites en bacterias puede regular la expresión de genes específicos o alterar su secuencia codificante dando como resultado variaciones antigénicas o de fase. Esto es particularmente favorable en bacterias patógenas cuando ocurre en loci de contingencia como forma de evadir las estrategias de defensa del hospedero 6,7. Como marcadores genéticos han sido ampliamente usados en estudios genéticos poblacionales debido a su elevado polimorfismo consecuencia de sus altas tasas de mutación, diversidad alélica, presentar codominancia y ser selectivamente neutros. Es también muy conocida su utilización en medicina forense para la identificación de personas y grado de parentesco.
Los microsatélites han sido identificados experimentalmente a partir de librerías genómicas de organismos de interés, inspeccionando miles de clones por hibridación con sondas de microsatélites. Además de su elevado costo, estos métodos contienen el sesgo propio de la composición de patrones de secuencia preseleccionados. Con la modernización y abaratamiento de las tecnologías de secuenciación, junto a las colaboraciones para el intercambio público de secuencias como GenBank, EMBL, DDBJ entre otras, los métodos de la bioinformática han tomado la supremacía surgiendo numerosas aplicaciones que implementan algoritmos orientados a este fin. No obstante, la propia dinámica de estas secuencias, sometidas a diferentes fuerzas evolutivas, sus roles particulares, así como el interés particular de los investigadores en unas u otras en dependencia de su composición, rasgos generales y fin biológico, ha hecho que estas aplicaciones bioinformáticas implementen criterios computacionales diferentes, y, por consiguiente, muestren variaciones en sus resultados 8,9. Por citar algunos ejemplos, encontramos aplicaciones que extienden su sistema de detección a regiones repetidas con periodicidades mayores (i.e. minisatélites y satélites); otras detectan sólo repetidos exactos o con mínimas variaciones definidas a priori; otras que emplean diccionarios preestablecidos de microsatélites o que detectan regiones de baja complejidad y luego las confirman empleando reglas establecidas. Aplicaciones como TRF, IMEx, START, SRF o TROLL 10-14 son ejemplos de software ampliamente utilizados para estos fines, e implementados con diversos criterios algorítmicos. La conclusión es que no existe una solución única, el espectro de aplicaciones se diversifica atendiendo a los tipos de SSR de interés y a los métodos computacionales empleados, haciéndose notar las diferencias en los resultados que retornan.
La aplicación que se presenta, MIDAS (MIcrosatellite Detection Assistant System), cumple con los siguientes principios generales: 1ro se detectan sólo microsatélites, exactos o inexactos (i.e. repetidos en tándem con patrones entre 1-8 pb, no compuestos ni complejos); 2do se detectan microsatélites exactos para luego extenderlos en caso de que sus flancos muestren alta similitud de secuencia con el patrón de repetición; 3ro la detección exacta es exhaustiva (i.e. se tienen en cuenta todos los posibles patrones que pudieran conformar un SSR); y 4to la extensión se hace por alineamiento local brindando una solución óptima de acuerdo a parámetros prefijados del alineamiento.
En la sección Métodos se describe en detalle la secuencia de pasos que sigue la aplicación, los fundamentos algorítmicos, la solución particular propuesta a los problemas de la detección inexacta de SSRs, y ejemplos de detección de SSRs exactos e inexactos.
En la sección Resultados, se expone y analiza la salida correspondiente a la detección de SSRs para el genoma de Salmonella entérica (subsp. entérica Serovar cubana str.). También se describen los parámetros y formatos de entrada y salida de la aplicación.
Métodos
El procedimiento inicia con la detección de una secuencia repetida exacta, es decir, sin sustituciones, inserciones o supresiones de bases. Para ello, en una primera etapa se implementa el autómata Aho-Crasick (AAC) que a partir de la construcción de un árbol de palabras encuentra todas las ocurrencias de éstas en un texto. En la implementación propuesta, se construye un árbol que contiene todas las palabras, de tamaños entre 1-8 nucleótidos (uno de los parámetros de la aplicación permite fijar el límite superior de este rango), formadas por combinaciones de las 4 bases nucleotídicas, y con la especificidad de que ellas mismas no constituyan secuencias repetidas (ej. aaaaa) y excluyendo las permutaciones cíclicas de ellas. ACC computa la búsqueda de estas ocurrencias eficientemente en tiempo proporcional al tamaño del texto y sin pre-procesamiento del mismo. Las ocurrencias de palabras iguales adyacentes son empalmadas y su posición registrada, estableciéndose los repetidos exactos o “semillas” de posibles repetidos inexactos. Con este paso, el algoritmo se comporta como cualquier otra aplicación que detecte repetidos exactos de forma exhaustiva y determinista (Fig. 1 (I)). Las limitaciones de los programas que detectan sólo estas secuencias son evidentes en cuanto al fin biológico que se persigue. Pensemos, solo a modo de ejemplo, en un repetido que tenga una simple modificación en una base, en cuyo caso el programa detectaría dos repetidos de la misma clase separados por una base, cuando en realidad constituían parte del mismo repetido. Este problema, que al generalizarse crea infinidad de situaciones menos triviales y complicadas de ejemplificar, constituye la principal motivación de la solución que se propone. En pocas palabras, se trata de detectar una “semilla” repetida exacta, con una cantidad de repeticiones razonable como para no ocurrir por simple azar, a partir de la cual detectar, si existiera, el repetido inexacto del cual ella forma parte. En caso de no existir dicha extensión aproximada o inexacta se reportaría el repetido exacto correspondiente, en este caso libre de ambigüedad.
La segunda etapa del algoritmo viene a solucionar el problema antes descrito. Se trata de buscar el posible candidato inexacto a partir de los flancos de la semilla exacta detectada previamente. En esta etapa se utiliza programación dinámica, es decir, el alineamiento local de la secuencia problema, incluidos los flancos, contra el patrón de repetición, empleando la eficiente técnica wraparound (WDP, Wraparound Dinamic Programming, por sus siglas en inglés) (Fig. 1(II), Fig. 2). Como es típico en los métodos de alineamiento de secuencias, la solución óptima es dependiente de los parámetros del alineamiento que definen las ponderaciones por coincidencias, sustituciones o inserciones/supresiones de bases, los cuales determinarán en última instancia el grado de conservación del microsatélite reportado.
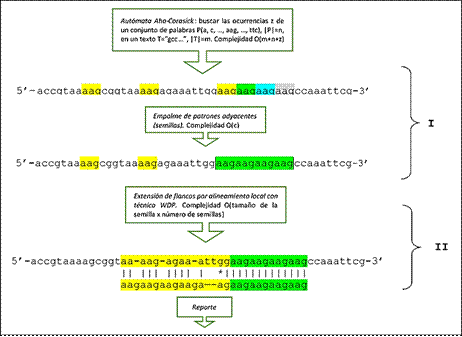
Fig. 1 Secuencia de pasos del algoritmo implementado en MIDAS en sus dos etapas fundamentales. (I) Detección de microsatélites exactos (semillas), usando árbol de palabras (Aho-Corasick) y posterior empalme de ocurrencias. (II) Extensión de semillas y detección del posible microsatélite inexacto empleando programación dinámica.

Fig. 2 Recurrencia que define el algoritmo de la segunda etapa en MIDAS. Es un alineamiento local clásico aplicando técnica wraparound. Basándose en la repetición del patrón, la ganancia en términos de tiempo y espacio se establece al permitir alinear la secuencia problema solo con el patrón o unidad repetida en el microsatélite. Match, mismatch (μ) e indel (δ) son los parámetros para coincidencias, sustituciones o inserción/supresión respectivamente.
Con respecto a la extensión per se hay dos problemáticas, que aparecen con poca explicitud en las aplicaciones reportadas por otros autores empleando alineamiento de secuencias, y que deben ser aclaradas y razonablemente solucionadas: 1ro ¿hasta dónde extender en los flancos de la secuencia para reportar el alineamiento? Cuando la secuencia problema es relativamente corta el problema desaparece si utilizamos la secuencia en su totalidad para buscar la sub-secuencia repetida local óptima. En la mayoría de los casos esto no es posible, pensemos por ejemplo en un cromosoma humano de más de 200 millones de pares de bases, con miles de microsatélites candidatos en diferentes regiones del genoma. La solución que presenta MIDAS es utilizar tamaños de flancos de 3 veces el tamaño de la semilla y si el alineamiento computado cubre más del 90 por ciento de la secuencia escogida se repite el proceso de extensión de flancos y se realinea la secuencia. De esta forma garantizamos que la región a extender para buscar el repetido inexacto sea dinámica y no excluya regiones donde se pueda seguir extendiendo. 2do ¿cómo evaluar si un alineamiento es lo suficiente adecuado para decidir seleccionarlo? Este problema es inherente a todos los métodos de alineamiento de secuencia y se ha afrontado de variadas formas en dependencia del contexto. En aplicaciones para detectar repetidos, algunos autores emplean el score o puntuación del alineamiento como criterio de selección (este es el caso de TRF). Este enfoque es en cierta medida arbitrario teniendo en cuenta que el score, si bien depende de los parámetros del alineamiento, también depende del tamaño del mismo, y se establece cierto sesgo que favorece a los SSR de mayor tamaño en detrimento de los más cortos, teniendo ambos igual importancia. En el caso de MIDAS este problema desaparece teniendo en cuenta que se parte de un SSR exacto y la extensión del mismo recaerá exclusivamente en los parámetros del alineamiento, en otras palabras, la aplicación no tiene la necesidad de escoger a priori un SSR estableciendo un valor de corte a partir de la puntuación del alineamiento.
Los parámetros del alineamiento son por defecto bastante restrictivos: (Match=2, Mismatch=-5, Indel=-5), aunque el usuario tiene la opción de usar otros esquemas de puntuación más relajados (4 en total) y luego podría depurar los SSRs a voluntad por inspección visual. La Figura 3 ejemplifica lo mencionado anteriormente con los resultados de la detección de dos SSR en un mismo genoma y con el mismo esquema de puntuación.

Fig. 3 Salida del programa para dos microsatélites situados en dos posiciones (separadas por 379821 pb) del genoma vibrio cholerae IEC224 (NC_016944). El patrón de secuencia es el mismo en ambos al igual que las unidades repetidas exactas (aag)4. La secuencia en verde es el SSR exacto, en amarillo la extensión inexacta y el resto son flancos. Los parámetros del alineamiento fueron (Match=2, Mismatch=-3, Indel=-3). En (I) el programa extendió a una guanina (g) coincidente en flanco izquierdo, sin embargo, en (II) hay una extensión mucho mayor que incluyen una sustitución y cinco inserciones o supresiones de bases. Notar que esta extensión quedaría reducida a una repetición exacta en caso de emplear parámetros más restrictivos, (ej. Match=2, Mismatch=-5, Indel=-5).
Resultados y Discusión
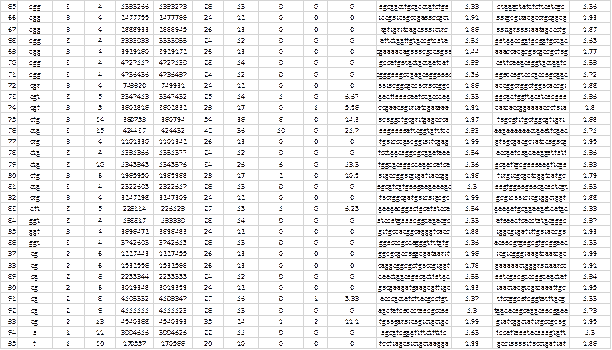
MIDAS se presenta en su versión 1.0 (binario) para plataforma Windows 32 y 64 bits (descargar midas_v1.0.zip del material suplementario) y su código fuente está escrito totalmente en C++ STL compatible, de modo que puede ser compilado para otras plataformas (Linux, Mac OS, etc.) empleando los compiladores y librerías adecuados. Es una aplicación de consola, ideal para hacer procesamientos por lotes (batch) y encadenar a otras aplicaciones (pipelines) mediante asignación de argumentos por línea de comandos. Los argumentos de la aplicación son tres: nombre de fichero (genoma a escanear en formato FASTA o GBFF ambos de tipo simple o multi-locus), tamaño máximo de la unidad repetida a escanear y esquema de parámetros del alineamiento para match, mismatch e indel, 4 en total, (2,-7,-7) (2,-5,-7) (2,-5,-5) (2,-3,-5). Como salidas MIDAS devuelve tres ficheros de tipo texto que tienen como nombre el fichero de entrada y las extensiones .xls, .dat y. mfaa (el .xls para abrir directamente con Excel u otra aplicación de hojas de cálculo). El fichero .xls (Fig. 5) tiene formato tabular y sus columnas son: Pattern (motivo), Length (tamaño de la unidad repetida), Copies (No. de copias), Start (posición inicial en el genoma), End (posición final en el genoma), Score (puntuación del alineamiento), Matches (bases coincidentes), Mismathces (bases no coincidentes), Indel (inserciones y supresiones de bases), Inaccuracy (% de inexactitud del repetido, medida de imperfección del mismo), 5' Flank (secuencia flanco al extremo 5´), 5'Entropy (entropía composicional del flanco 5´), 3' Flank (secuencia flanco al extremo 3´), 3'Entropy (entropía composicional del flanco 3´). Algunos autores de software para la detección de repetidos en tándem reportan la entropía composicional de la región repetida, siendo esta obvia y mayoritariamente baja. Lo que permite que un microsatélite pueda utilizarse como marcador genético es las variaciones en el número de copias y los flancos que permiten su amplificación en técnicas de PCR. MIDAS reporta la entropía de los flancos, candidatos para diseñar los cebadores en técnica de PCR, dando una medida de cuán informativos y únicos pueden ser éstos en el genoma.
El fichero con extensión .dat presenta en forma no tabular los datos anteriores y permite visualizar el alineamiento de secuencia. Por último, el fichero con extensión. mfaa presenta los microsatélites detectados en formato multi-fasta, en el cual la región del repetido está marcada en minúscula y los flancos en mayúscula. Este formato permite hacer procesamiento por lotes (batch) con blastn (opción de enmascaramiento explícito de sub-secuencias), para la búsqueda de candidatos polimórficos intra- e inter- especies (Fig. 4). El encabezado de este fichero presenta información como el número de acceso del GenBank, el motivo y las posiciones en el genoma.

Fig. 4 Formato del fichero con extensión. mfaa (multi-fasta con la región repetida marcada con letra minúscula).
El genoma de Salmonella enterica (subsp. enterica Serovar cubana str., código de acceso NC_021818) tomado del repositorio del NCBI (https://www.ncbi.nlm.nih.gov/), fue escaneado con MIDAS y los resultados se muestran en Fig. 5. Este genoma presenta 4,977,480 pares de bases (pb) y el tiempo de cómputo, incluida la creación de los ficheros de salida, fue menor de 3 segundos. Un total de 95 SSRs fueron detectados, 2 hexa-, 14 tetra-, 70 tri-, 7 di- y 2 mono-nucleótidos (cantidad y unidad repetida respectivamente). Los parámetros de detección fueron: unidad repetida <=6 y Match=2, Mismatch=-3, Indel=-3 (esquema tipo 4 como parámetros del alineamiento para la fase de extensión). Es notable el porcentaje de SSRs con tri-nucleótidos como unidad repetida (74%), lo que hace sospechar de su localización en regiones codificantes fundamentalmente, a pesar de que en los genomas bacterianos predominan estas regiones. Los números de copia muestran un rango de 3 a 15, con media 5.8, para un coeficiente de variación de 50 %, resaltando entre estos los SSRs 1 y 2 (de hexa-nucleótidos con 14 copias), 37, 75 y 76 (de tri-nucleótidos con 13, 14 y 15 copias respectivamente) y el 93 (de di-nucleótidos con 13 copias).
La entropía composicional media de los flancos 5´ y 3´ es de 1.86 y 1.88 respectivamente, los cuales se pueden considerar elevadas por su cercanía a 2 (valor máximo). Entre estas destaca curiosamente el flanco 3´del SSR No. 23 y el flanco 5´del SSr No. 30, ambos con entropía composicional máxima.
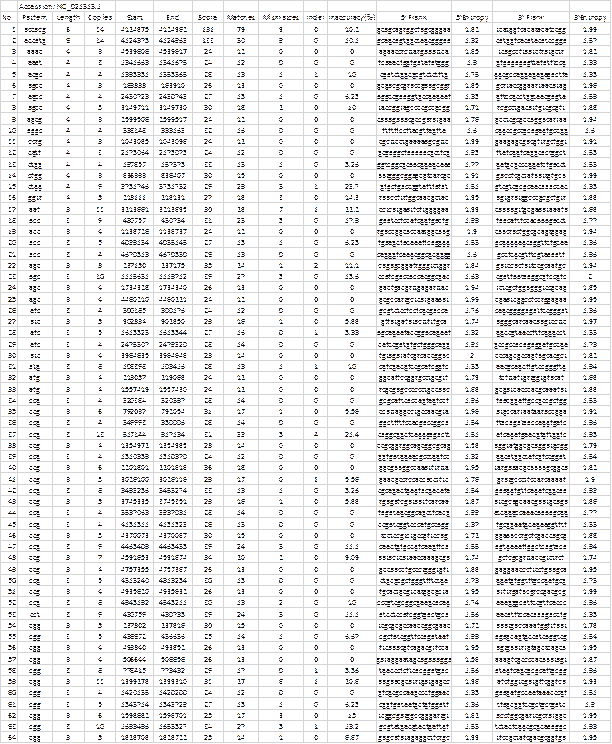
Fig. 5 Resultados de la detección de SSRs en el genoma de Salmonella enterica (subsp. enterica Serovar cubana str., código de acceso NC_021818). Esta representación es la que muestra el fichero de salida con extensión .xls.
Conclusiones
Se presenta la aplicación MIDAS para la detección de microsatélites (SSRs) exactos e inexactos. El algoritmo es totalmente combinatorio y tiene dos etapas o procedimientos generales: 1ra detección de SSRs exactos por técnica de reconocimiento de patrones de texto exactos y 2da extensión de los mismos mediante técnica de programación dinámica. Se muestran los resultados, y un breve análisis, de la detección de estas secuencias en el genoma de Salmonella entérica (subsp. entérica Serovar cubana). La aplicación es eficiente e intuitiva, presentando tiempos de ejecución bajos (4,977,480 pb en 3 seg.) y una cantidad mínima de parámetros de entrada lo cual lo hace más asequible para el usuario. Presenta formatos de salida descriptivos, tabulados y bioinformáticos que permiten una fácil y muy completa visualización para el análisis de los resultados, permitiendo también el encadenamiento de éstos con otras aplicaciones, por ejemplo, para extracción de rasgos anotados en otros repositorios o detección de polimorfismos mediante búsquedas extensivas de tipo BLAST.

