










 Custom services
Custom services Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La enfermedad de Rendu-Osler-Weber (ROW), también conocido como telangiectasia hemorrágica hereditaria (THH), es una enfermedad genética de herencia autosómica dominante con penetrancia incompleta,1,2 con una incidencia de 1-2 por cada 100 000 personas, distribuidas homogéneamente por raza y sexo;1 se estima una prevalencia a nivel mundial de 1:5000 y 10 000 personas. Afecta por igual a ambos sexos y los síntomas se inician habitualmente entre los 20 y 40 años,3 pero se considera que la enfermedad está infradiagnosticada.4 El cuadro clínico se caracteriza por la presencia de epistaxis recurrentes y espontáneas, de inicio en la edad infantil, instauración progresiva de telangiectasias mucocutáneas en labios, cavidad bucal, nariz y dedos, asimismo el desarrollo de malformaciones arteriovenosas (MAV) viscerales, fundamentalmente pulmonares, gastrointestinales, hepáticas y del sistema nervioso central.1,2,3,4,5
Esta enfermedad se describe por primera vez en el siglo xix como un trastorno familiar que causaba sangramiento nasal e intestinal y se asociaba a estructuras vasculares anormales.6,7 Después de la descripción de H. Rendu,8W. Osler9 y F. Weber,10 la enfermedad atrajo la atención de la comunidad médica, lo cual resultó en el epónimo de síndrome de ROW, aunque el término THH acuñado por F.M. Hanes es más notificado.11
Típicamente, las formas clínicas y el debut de esta enfermedad se asocian a los órganos y tejidos que se afectan con mayor frecuencia: telangiectasias en mucosas y en piel, epistaxis, sangramiento gastrointestinal, pulmonar e intracerebral.3,4 En contraste, el caso clínico que se presenta se caracteriza porque las primeras manifestaciones clínicas que motivaron la consulta médica fueron crisis de dolores e inflamación ósea en el miembro superior derecho, lo cual es inusual y se inscribe como un elemento de novedad en la enfermedad ROW. Es el objetivo de esta publicación exponer un caso de THH con forma de presentación atípica en una adolescente y así, contribuir a la trasmisión del conocimiento sobre una entidad en la cual el pronóstico para la supervivencia de los pacientes será favorable si las complicaciones se diagnostican y se tratan oportunamente.12
Presentación del caso
Paciente femenina, de 16 años de edad, con antecedentes previos de salud que refirió haber tenido dolor y leve inflamación en la zona distal del radio derecho a mediados del 2019, que se alivió espontáneamente y sin dejar secuelas. Seis meses más tarde se repitió este episodio de dolor e inflamación más intensos, en la misma zona, notándose un aumento de volumen duro no movible en la región del antebrazo derecho, lo que motivó su consulta médica en el Servicio de Pediatría de su área de salud. Allí se le realizaron varios estudios imagenológicos en la zona afectada constatando que el aumento de volumen era a expensas del radio; además había un aumento de grosor sin romper la cortical, por lo que se decidió realizar biopsia de músculo y tejido óseo. Durante la realización de este procedimiento llamó la atención el sangramiento profuso en la zona de incisión que causó incluso la disminución de los niveles de hemoglobina en la paciente; pero el resultado de dicho estudio no arrojó un diagnóstico preciso. Durante el chequeo preoperatorio se realizó radiología de pulmón que informó nódulos pulmonares, lo cual se corroboró por tomografía axial computarizada (TAC) monocorte y se decidió realizar biopsia pulmonar, con lo cual no estuvo de acuerdo la madre y se trasladó a nuestro centro en el que ingresó en el Servicio de Reumatología Pediátrica en el Hospital Juan Manuel Márquez como sospecha de enfermedad granulomatosa.
Al ingreso se constató al examen físico una masa tumoral no dolorosa en la zona distal del radio derecho (Fig. 1A) que se presentó como un aumento de volumen (la madre refirió que aumentó su tamaño después de la biopsia), y además se apreció que los dedos de las manos y los pies tenían la morfología descrita como “dedos en palillo de tambor” (Fig. 1B).

Fig. 1 - A. Estudio radiológico de ambos brazos. Reacción perióstica densa con engrosamiento cortical medial del tercio distal de la diáfisis del radio derecho, asociada a discreto incremento de la densidad radiográfica de las partes blandas. B. Morfología característica del denominado “dedos de palillo de tambor”.
Adicionalmente, los resultados del análisis gasométrico evidenciaron una saturación de oxígeno del 90 %.
Durante la hospitalización se realizaron análisis de laboratorio clínico, de anatomía patológica y estudios de imágenes, cuyos resultados se resumen en la tabla 1. Además, el interrogatorio y el examen físico revelaron antecedentes de epistaxis y presencia de telangiectasias en el tórax y la mucosa bucal. También se efectuaron interconsultas con otros especialistas, de lo cual se referirán solamente las observaciones relevantes concernientes a esta enfermedad.
Tabla 1 - Resumen de los estudios realizados durante la hospitalización de la paciente
| Análisis de laboratorio clínico | Hemoquímica, leucograma, etc. | Sin alteraciones | - |
| Gasometría | Saturación de oxígeno del 90 % | - | |
| Ultrasonido abdominal | Sin alteraciones en hemiabdomen superior | - | |
| Anatomía patológica | Biopsia de hueso | Fragmento de tejido esponjoso sin alteraciones, no tumoral, y fragmento de tejido adiposo muy vascularizado, no tumoral | - |
| Survey óseo | Rayos X de cráneo (frontal) | Sin alteraciones | - |
| Rayos X de miembros superiores | Reacción perióstica densa con engrosamiento cortical medial del tercio distal de la diáfisis del radio derecho | Fig. 1 A | |
| Rayos X de miembros inferiores | Fibroma no osificante en la tibia derecha | Fig. 2A | |
| Rayos X de tórax (anteroposterior y lateral) | Imágenes nodulares en la base pulmonar derecha | Fig. 2B | |
| TAC multicorte contrastada | Cráneo | Pólipo en el seno maxilar derecho e hipertrofia de cornetes. No se definieron malformaciones vasculares. No se observaron alteraciones craneoencefálicas ni alteraciones óseas | Fig. 3 |
| Tórax | Lesiones nodulares múltiples de tamaño variable con gruesos vasos aferentes y eferentes, arteriales y venosos, sugestivos de malformación arteriovenosa | Fig. 4A Fig. 4B | |
| Abdomen | Focos de realce parenquimatoso posiblemente relacionados con etiología vascular en el hígado | Fig. 5 | |
| Miembro superior derecho | Engrosamiento cortical de contornos ondulantes y bien definidos, diafisiario, en el radio derecho y con incremento de la densidad de partes blandas | Fig. 6 | |
Fuente: Historia clínica.
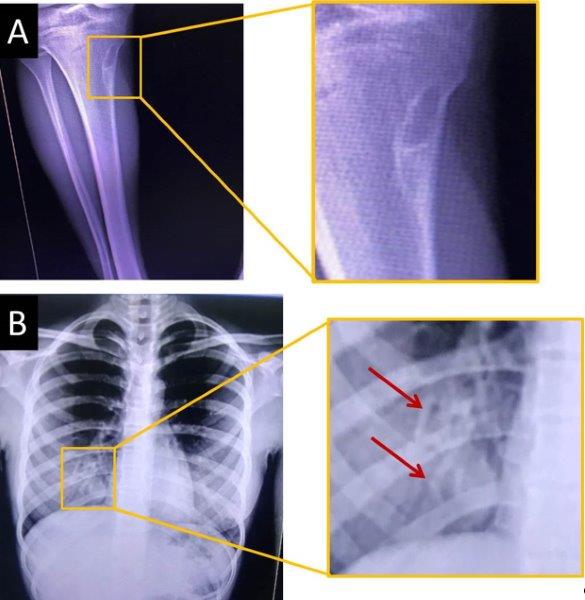
Fig. 2 - A. Imagen osteolítica metafisodiafisaria del tercio proximal de la tibia derecha, con base cortical y contorno esclerótico fino bien definido en posible relación con fibroma no osificante que mide 31 mm x 9 mm (recuadro superior). B. Rayos X de tórax: imágenes nodulares en la base pulmonar derecha (recuadro inferior y flechas).
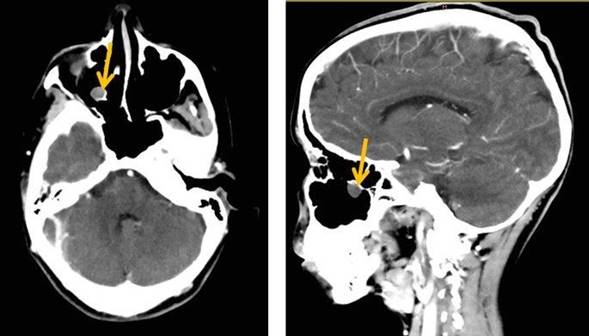
Fig. 3 -Tomografía axial computarizada de cráneo. Cortes axiales a 1,5 mm con reconstrucción multiplanar (0,44 mm). Corte transversal (izquierdo) y corte sagital (derecho). Se observa un pólipo en el seno maxilar derecho (flecha) que mide 15 mm craneocaudal, 12 mm anteroposterior y 10 mm transversal.

Fig. 4 - A. Tomografía axial computarizada contrastada de tórax. Reconstrucción en 3D de ambos pulmones. Cortes axiales a 2 mm con MPR a 0,98mm. En ambos campos pulmonares se observaron lesiones nodulares múltiples de tamaño variable, las mayores bibasales. Dimensiones de los nódulos: en el lóbulo medio del pulmón derecho: 24 x 20 mm; en el lóbulo inferior del pulmón izquierdo: 21 x 19 mm. B. Reconstrucción focalizada en 3D del TAC de tórax. En el estudio contrastado las lesiones se muestran hipercaptantes, con densidades medias que oscilan entre 168-191 UH, con gruesos vasos aferentes y eferentes, arteriales y venosos, sugestivos de malformación arteriovenosa en el campo pulmonar izquierdo.
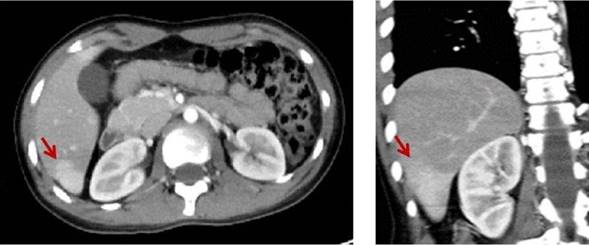
Fig. 5 -Tomografía axial computarizada de abdomen. Corte transversal (izquierdo) y corte coronal (derecho). Se observan dos zonas de realce parenquimatoso en el hígado (flechas) que miden 30 x 24 mm y 16 x 8 mm, con densidades medias entre 156-170 UH, posiblemente asociadas a lesiones focales de etiología vascular.
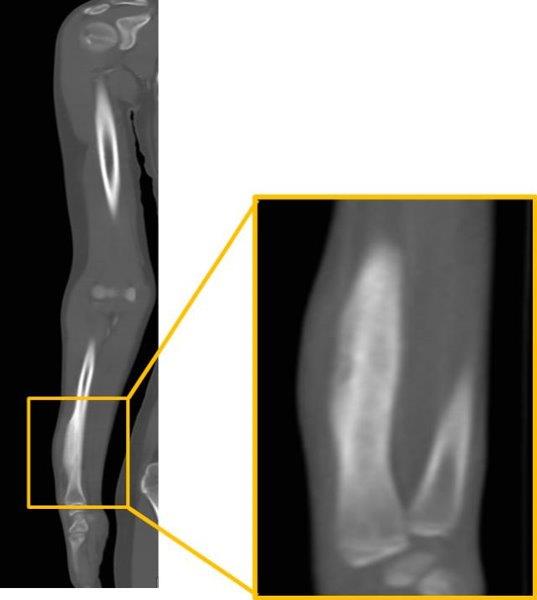
Fig. 6 -Tomografía axial computarizada contrastada de antebrazo derecho. Izquierda: Se observa engrosamiento cortical de 12 mm, de contornos ondulantes y bien definidos, diafisiario, con incremento de la densidad de partes blandas (recuadro).
Interconsultas
Se consultaron otras especialidades médicas, que aportaron lo siguiente:
Oftalmología: El fondo de ojo evidenció un nódulo en el ojo izquierdo y hemorragia en el ojo derecho, las cuales se asociaron a posibles fístulas arteriovenosas.
Dermatología: El examen de la piel evidenció dos fibromas cutáneos puntiformes.
Genética: Como antecedente patológico familiar se refirió una tía paterna que sufría epistaxis frecuente, no estudiada. Se diagnosticó la enfermedad de Rendu-Osler-Weber de acuerdo con el antecedente de sangrado nasal frecuente, presencia de telangiectasias dentro de la mucosa bucal y malformaciones vasculares en el pulmón.
Al alta hospitalaria, la paciente se hallaba estable, sin complicaciones. Se le recomendó el seguimiento en el hospital de su provincia, fundamentalmente por la especialidad de Neumología, por ser los pulmones los órganos más afectados.
Discusión
La enfermedad de ROW ha permanecido durante mucho tiempo ignorada en la literatura médica, probablemente debido al subregistro de esta entidad.13 Es una enfermedad rara y multisistémica, con un amplio espectro de formas de presentación clínica y un riesgo sustancial de complicaciones. Habitualmente los clínicos no tienen presente esta enfermedad y con frecuencia no la reconocen a tiempo.4
El diagnóstico es clínico y se basa en los criterios de Curaçao. El diagnóstico es definitivo con 3 de 4 criterios y es posible con 2 de 4 (Tabla 2).14
Tabla 2 -Criterios de Curaçao para el diagnóstico clínico de telangiectasia hemorrágica hereditaria
| Epistaxis | Espontánea o recurrente |
| Telangiectasias | Múltiples y en sitios característicos: labios, cavidad bucal, dedos y nariz |
| Lesiones viscerales | Telangiectasias gastrointestinales, pulmonares, hepáticas, malformación arteriovenosa cerebrales o espinales |
| Antecedentes familiares | Un familiar de 1.° grado con telangiectasia hemorrágica hereditaria acorde con los criterios expuestos |
Aunque no existen estudios que evalúen la sensibilidad y especificidad de estos criterios, son ampliamente aceptados en la práctica clínica.
El diagnóstico genético es posible, pero en el 15-20 % de los casos no se detecta mutación alguna. También se ha planteado la posibilidad de un diagnóstico serológico, pero se encuentra disponible en pocos laboratorios y se emplea principalmente con fines de investigación. Se plantea su utilidad en pacientes jóvenes o pediátricos en quienes se sospeche la enfermedad, pero no cumplan los criterios clínicos, en particular sin telangiectasias visibles ni epistaxis, teniendo en cuenta que estas manifestaciones aparecen por lo general hacia la cuarta década. En el caso de no estar disponible el estudio genético, una alternativa para el diagnóstico en este último grupo es la búsqueda de MAV viscerales.13,14
Los estudios para las MAV comprenden: ecosonografía Doppler visceral, tomografía de alta resolución combinada con reconstrucción 3D, resonancia magnética nuclear, endoscopía y angiografía digital, que es el método diagnóstico de elección por su alta sensibilidad y especificidad.
Este caso tuvo la particularidad de que la deformidad ósea que presentó la paciente en la región del antebrazo dio paso al diagnóstico de esta enfermedad. Inicialmente se interpretó que la periostitis en la zona del radio derecho estuviera relacionada con sangramiento local por malformación vascular en dicha zona, dado el antecedente de sangrado profuso durante la biopsia realizada que pudo contribuir además al aumento de dicha reacción perióstica. Aunque los estudios realizados no permitieron respaldar esta teoría, se conoce que las MAV se pueden presentar en cualquier parte de la economía. Lo anterior resulta atípico, pues al alcance de la revisión bibliográfica realizada para documentar este estudio no se ha encontrado ningún caso similar.
Se ha informado que en el 70 % de los individuos afectados con la enfermedad de ROW, esta comienza a manifestarse alrededor de los 16 años,13,14 lo cual coincide con el comienzo de la enfermedad en la paciente de este estudio de caso.
La imagen sugestiva de fibroma óseo no osificante se considera un hallazgo radiográfico que se puede presentar a esta edad (entre los 10 y 20 años, coincidiendo con la paciente) y no guarda relación con la enfermedad de base.15
La afectación ocular en la THH se incluye en el cuadro clínico desde las primeras publicaciones acerca de la enfermedad. La afectación más frecuente es la presencia de telangestasias en la conjuntiva, ya descrito por Chauffaerd en 1896 y Weber en 1907. La afectación intraocular es más rara, y en varios casos de los descritos en pacientes con alteraciones retinianas se presentó como oclusión de la rama venosa retiniana.16
Las MAV pulmonares son comunicaciones directas anormales entre arterias y venas pulmonares. Aproximadamente el 80-90 % de estas corresponden a la enfermedad de ROW, aunque solo del 15-35 % de los pacientes que lo padecen presentan dichas malformaciones. Por lo general, son múltiples, bilaterales y se ubican en los lóbulos inferiores posteriores,14 como en este caso. Esos hallazgos demandarán un seguimiento médico sistemático del enfermo, ya que habitualmente se asocian con complicaciones graves tales como accidentes cerebrovasculares, hemoptisis masiva y hemotórax espontáneo,17,18 específicamente durante el embarazo.19
Las MAV pulmonares proporcionan una comunicación capilar libre entre la circulación pulmonar y sistémica que tiene tres consecuencias deletéreas: (a) que la sangre de las arterias pulmonares que pasa a través de estas derivaciones no pueda oxigenarse, y conduce a la hipoxemia (presente en este caso, con una saturación de oxígeno del 90 %), (b) que la ausencia del filtro que representa el lecho capilar normal permita que material particulado alcance la circulación sistémica donde impactará en otros lechos capilares con secuelas clínicas particularmente en la circulación cerebral, y (c) que la fragilidad de los vasos sanguíneos puede conducir a hemorragias dentro de los bronquios (asociado a hemoptisis) o en la cavidad pleural (asociado a hemotórax).16
En la mayoría de los pacientes con la enfermedad de ROW que exhiben lesiones hepáticas, estas cursan de manera silente y pueden ir desde lesiones macroscópicas hasta microscópicas.7 Se presentan desde pequeñas telangiectasias hasta grandes MAV. Aunque anteriormente se pensaba que era poco frecuente, entre el 8-30 %, hoy se sabe que la afectación hepática es muy frecuente y llega al 74 % de los pacientes afectados. Sin embargo, solo un 8 % tiene shunts hepáticos sintomáticos y la mayoría de ellos son mujeres.13 En la paciente sujeto de este estudio se constataron lesiones hepáticas relacionadas con posibles telangiectasias las cuales hasta el momento no han tenido implicaciones funcionales. Cuando las malformaciones vasculares hepáticas tienen trascendencia clínica, estas se presentan como insuficiencia cardiaca, hipertensión portal o enfermedad vascular, lo cual refleja patrones diferentes de implicación vascular.2,20
Todos los hallazgos imagenológicos en este caso condicionaron la atención especializada multidisciplinar enfocada a prevenir oportunamente cualquiera de las complicaciones esperadas por el grado de implicación de diferentes órganos y sistemas. Particularmente en esta paciente, de por vida deberá implementarse el tratamiento profiláctico con antibióticos de amplio espectro durante intervenciones estomatológicas u otras intervenciones quirúrgicas con el fin de reducir el riesgo de absceso cerebral. En los próximos años, durante su vida reproductiva, esta paciente deberá recibir asesoramiento genético y su descendencia deberá investigarse con el propósito de diagnosticar precozmente si padecerá la enfermedad.
Conclusión
Al considerar lo atípico del debut de la enfermedad de ROW, la descripción pudiera ser un referente de utilidad para médicos generales y especialistas que en su práctica habitual pueden encontrar casos similares. Hacer un diagnóstico más expedito y precoz permitiría que esos pacientes reciban prontas alternativas terapéuticas y recomendaciones de estilos de vida compatibles con el desarrollo de una vida lo más normal posible.

