










 Custom services
Custom services
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introducción
Los Trypanosomatidae forman una familia de parásitos eucariotas unicelulares del orden Trypanosomatida, clase Kinetoplastida, phylum Euglenozoa, supergrupo Excavata. Varios patógenos humanos son vectores de las especies de los géneros Leishmania y Trypanosoma. Los ciclos de vida de estos organismos resultan ser muy complejos, moviéndose constantemente entre insectos y hospedadores mamíferos.1 Las especies Leishmania spp., Trypanosoma cruzi y Trypanosoma brucei son los agentes causantes de la leishmaniasis, la tripanosomiasis americana (enfermedad de Chagas) y la tripanosomiasis africana humana (enfermedad del sueño), respectivamente.2
Las leishmaniasis son un grupo diverso de enfermedades tropicales desatendidas que afectan humanos en 98 países, y son causadas por al menos veinte especies de los parásitos protozoarios del género Leishmania. Las manifestaciones de la enfermedad van desde la leishmaniasis cutánea autocurativa hasta la leishmaniasis mucocutánea mutilante o leishmaniasis cutánea difusa 3 y leishmaniasis visceral potencialmente mortal.4 La Organización Mundial de la Salud (OMS) estima que hay aproximadamente 0,2-0,4 millones de nuevos casos de leishmaniasis visceral y 0,7-1,2 millones de nuevos casos de leishmaniasis cutánea por año.5 La enfermedad no ocupa un lugar destacado en la agenda política o de investigación de muchos países, ya que normalmente afecta a los más pobres entre los pobres.6
La enfermedad llega a la población humana cuando el humano y los huéspedes reservorios comparten el mismo entorno.7 Las especies de leishmania se transmiten en un ciclo zoonótico, con perros o roedores como reservorio animal, pero otras son estrictamente antroponóticas.8
Las alternativas terapéuticas más usuales incluyen sales pentavalentes de antimonio, desoxicolato de anfotericina B y anfotericina B liposomal, paromomicina, pentamidina y azoles. Desde el inicio del tratamiento con antimoniales pentavalentes, se ha observado que los enfermos se resisten al tratamiento debido a su gran toxicidad y efectos secundarios; a su vez, se desarrollan cepas más resistentes del parásito. La mejora del desoxicolato de anfotericina B en forma liposomal redujo los efectos secundarios; sin embargo, el impacto financiero en las regiones pobres es preocupante. Se han desarrollado otros fármacos, entre ellos la miltefosina; sin embargo, existen cepas resistentes, y en la forma tegumentaria de la enfermedad, la respuesta varía según la especie del parásito. En este sentido, es importante desarrollar nuevas estrategias de tratamiento para la población afectada. (9-10
La enfermedad de Chagas es una enfermedad tropical desatendida con alta prevalencia, morbilidad y mortalidad significativas. Las estimaciones de la carga de morbilidad de la Organización Mundial de la Salud colocan a la enfermedad de Chagas en el primer lugar entre las enfermedades parasitarias en las Américas. La transmisión ocurre cuando las heces de los insectos infectados contaminan el sitio de la picadura o las membranas mucosas intactas.11 Sin embargo, durante la última década se han reportado numerosos casos de infección por T. cruzi por vía oral, debido a la contaminación de alimentos con heces de triatominos silvestres.12
El tratamiento de La enfermedad de Chagas todavía se basa en solo dos medicamentos, nifurtimox y benznidazol, los cuales fueron desarrollados en la década de 1970 13 y tienen una eficacia limitada en la fase crónica tardía, causan efectos secundarios frecuentes y tienen resistencia a los medicamentos evolucionada.14
La tripanosomiasis africana humana, o enfermedad del sueño, es una enfermedad parasitaria transmitida por la mosca tsetsé (Glossina spp.) y tiene una prevalencia fluctuante en el África subsahariana. La enfermedad es causada por dos subespecies de Trypanosoma brucei: T. b. gambiense (que causa la forma crónica más prevalente de la enfermedad) y T. b. rhodesiense (una subespecie mucho más virulenta y menos común). Ambas formas de la enfermedad progresan a través de una etapa inicial y temprana que involucra la infección de los sistemas sanguíneo y linfático, evolucionando hacia una infección del sistema nervioso central (SNC) en etapa tardía donde surgen los síntomas característicos: interrupción del sueño, confusión, coma y, si no se trata, muerte.15
Para la tripanosomiasis africana humana, la mayoría de los pacientes se diagnostican durante la etapa de afectación del SNC, se requieren fármacos que traspasen la barrera hematoencefálica. Esto ha dado lugar a una farmacopea bifurcada, en la que la infección del torrente sanguíneo se trata con los fármacos que no penetran en el cerebro suramina y pentamidina, mientras que la infección del SNC se ha tratado con eflornitina (con o sin nifurtimox) o melarsoprol (un agente tóxico que contiene arsénico) y que tiene una tasa de mortalidad del 5,7 %. Este último fármaco, hasta la reciente implementación de la terapia de combinación nifurtimox/eflornitina (NECT), era el único terapéutico disponible para las infecciones por T. b. gambiense y ahora está restringido al tratamiento del estadio 2 de la infección por T. b. rhodesiense.15
Todos estos medicamentos adolecen de limitaciones importantes que van desde la eficacia específica de la etapa hasta una toxicidad significativa, así como la necesidad de administración parenteral (intravenosa para suramina, melarsoprol y eflornitina e intramuscular para pentamidina), lo que plantea desafíos prácticos en las zonas rurales de África.16
Un sexto fármaco, el fexinidazol, concluyó recientemente con éxito los ensayos clínicos. Los resultados de los ensayos clínicos con fexinidazol administrado por vía oral demostraron que es seguro y eficaz contra T. b. gambiense, lo que lo convierte en el primer nuevo tratamiento terapéutico en tres décadas, así como la primera monoterapia oral tanto en etapa 1 como en etapa 2.17 No obstante, la resistencia se induce fácilmente in vitro y el fexinidazol muestra resistencia cruzada con el nifurtimox. Además, este fármaco requiere un régimen de tratamiento de alta carga de píldoras, lo que subraya que los esfuerzos de investigación para el descubrimiento de nuevas terapias para tratar esta enfermedad tropical desatendida siguen siendo de gran interés.16
No es de extrañar, que para estas tres enfermedades, la búsqueda de alternativas farmacológicas sea un reto para un gran número de investigadores del mundo.9,18,19 La proteína Cyp51 del citrocromo P450 en la actualidad es una de las dianas más empleadas para el diseño de fármacos. El Cyp51 es empleado comúnmente como diana para el diseño de antifúngicos.20-21 Varios triazoles utilizados como fungicidas, exhiben actividad tripanocida tanto in vitro como in vivo. El mecanismo de acción de tales fármacos se basa sobre la inhibición de la biosíntesis de ergosterol que afecta la viabilidad y el crecimiento celular.18
Para vencer el reto del diseño de nuevos candidatos a fármacos, existe una gran cantidad de técnicas, entre las que se destacan las técnicas computacionales como QSAR, 22,23 acoplamiento molecular (molecular docking) 24-26 y/o dinámica molecular.27,28
A lo largo de su actividad, el grupo de investigación de Compuestos Bioactivos y Química Sostenible de la Universidad de Oriente ha diseñado un buen número de imidazoles sustituidos con potencial como el imidazol 4,5-difurilsustituido, dada su probada actividad antifúngica de interés agrícola. A su vez, estudios para evaluar la probable actividad antileishmania mediante el acoplamiento molecular de imidazoles 2-aril, 4,5-difuril sustituidos con diferentes blancos terapéuticos 29, orientan hacia el estudio de los imidazoles 2-[5-(4-nitrofenil)furil]-4,5-difurilimidazol y 2-[5-(4-carboxifenil)furil]-4,5-difurilimidazol. Estos resultados sugieren que se debe continuar el estudio de imidazoles 4,5-difurilsustituidos como potenciales agentes antimicrobianos.
Recientemente, en el grupo de investigación se han sintetizado imidazoles 2,4,5-triaril sustituidos y 1,2,4,5-tetraaril sustituidos donde, en su mayoría, las posiciones 4 y 5 son ocupadas por el fenilo y en menor medida por el grupo 2-furilo. Los mismos no habían sido estudiados como posibles agentes terapéuticos. Así, se han realizado estudios de acoplamiento molecular explorando su interacción con diferentes blancos terapéuticos de Mycobacterium tuberculosis30, de la proteína oncogénica Bcr-Abl Tirosina Kinasa 31, y estudios combinados in vitro e in silico por acoplamiento molecular de su actividad antiprotozoaria.1,32
Tomando en consideración estos elementos, el objetivo del presente trabajo es realizar un estudio comparativo in sillico, empleando el método de acoplamiento molecular, que permita evaluar la diferencia de emplear sustituyentes fenil o 2-furil en las posiciones 4 y 5 de imidazoles tetraarilsustituidos y triarilsustituidos como potenciales antiprotozoarios contra Leishmania spp., Trypanosoma cruzi y Trypanosoma brucei.
Materiales y métodos
Recursos computacionales
Los cálculos fueron realizados basados en los recursos computacionales del HPC de la Universidad de Oriente, clúster de tipo especializado. Se utilizó el sistema operativo Linux distribución Debian 8.0.
Modelación molecular de proteínas y ligandos para el acoplamiento molecular
Para la representación de las estructuras de los imidazoles sustituidos (figura 1), se empleó el programa ChemDraw Professional 15.0.33 El programa Chem3D 15.0, 33 se utilizó para realizar una optimización previa de las geometrías de los mismos, utilizando el método de mecánica molecular con campo de fuerza MMFF94 (en inglés Merck Molecular Force Field).
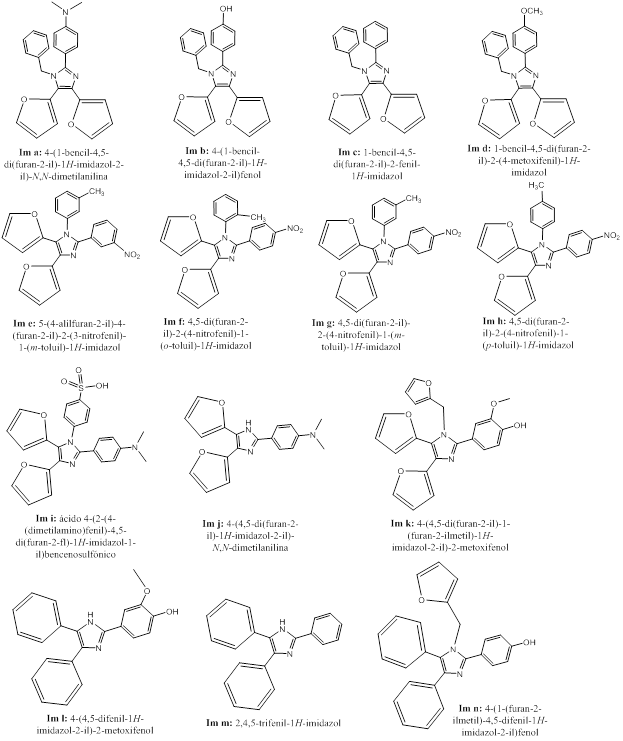
Para la modelación molecular de las proteínas se obtuvieron sus estructuras tridimensionales en formato .pdb, provenientes de la Base de Datos de Proteínas (en inglés Protein Data Bank), la cual es de acceso gratuito. Como estas estructuras son obtenidas generalmente por difracción de rayos X, con el software UCSF Chimera versión 1.10.227 34 se identificaron todos los residuos provenientes de la cristalización de la macromolécula que no pertenecen como tal a las proteínas en su estado natural y podían interferir con sus centros activos y, por lo tanto, con la interpretación de los resultados.
El estudio de acoplamiento molecular se llevó a cabo a través de los programas AutoDock 4.2 y AutoDock Tools 1.5.6. Se utilizó el método de cálculo de carga de Gasteiger 35 y se agregaron cargas parciales a los átomos del ligando antes del acoplamiento, a través de Autodock Tools. La visualización bidimensional de las interacciones hidrofóbicas y por puente de hidrógeno de los complejos, se efectuó por el programa Ligplot 36 y UCSF Chimera versión 1.10.227.34
Las proteínas seleccionadas para este estudio corresponden al Citocromo P450, lanosterol 14α demetilasa CYP51, de las especies T. cruzi, T. brucei y L. infantum. Para el caso del CYP51 de T. cruzi, esta proteína se encuentra formando un complejo con el inhibidor VNF ((4-(4-clorofenil)-N-[2-(1H-imidazol-1-il)-1-feniletil]benzamida) y su PDBID es 3KSW 37 con una resolución de 3.05 Å. El CYP51 de T. brucei se encuentra unido al (S)-N-(3-(1H-indol-3-il)-1-oxo-1-(piridin-4-ilamino)propan-2-il)-3,3′-difluoro-(1,1′-bifenil)-4-carboxamida con PDBID: 4BJK 38 y resolución de 2.67 Å. Para L. infantum la proteína CYP51 se encuentra formando un complejo con el fluconazol, su PDBID es 3L4D 39 y su resolución de 2.75 Å.
En las estructuras de las proteínas 3L4D y 4BJK se seleccionó la cadena A para la realización del acoplamiento molecular. La estructura de la 3KSW presenta una sola cadena.
Elección de parámetros para el acoplamiento molecular
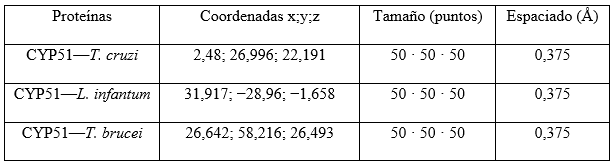
AutoDock requiere de la realización de un pre-cálculo de los mapas grid, uno para cada tipo de átomo presente en el ligando y ubicar, a su vez a dicho ligando, alrededor de la región de interés (sitio activo) en la macromolécula. Para lograr esto se requiere suministrar una serie de parámetros afines. En la tabla 1 se muestran los parámetros elegidos para cada una de las proteínas estudiadas.
Tabla 1 Parámetros para la realización del precálculo de los mapas grid y la ubicación del ligando alrededor de la región del sitio activo de la macromolécula
El Algoritmo Genético Lamarckiano (LGA) fue utilizado para explorar el mejor espacio conformacional para el ligando, con 50 corridas para cada uno de los acoplamientos. Se ajustó el cálculo a un número de evaluaciones de la energía de 2,5 · 106.
Resultados y discusión
Análisis del acoplamiento molecular de los imidazoles con las proteínas estudiadas
A continuación se evalúa el efecto de la sustitución en las posiciones 4 y 5 del imidazol por grupos fenilo o 2-furilo. Para ello se toman como muestra catorce imidazoles sustituidos y se analiza su interacción con las proteínas CYP51 de T. cruzi, T. brucei y L. infantum. Para ello se tomó como referencia lo reportado por Rojas y colaboradores 1, donde 11 imidazoles tetrarilsustituidos y 3 triarilsustituidos fueron sintetizados, determinadas sus actividades antiprotozoarias in vitro y analizada su interación con enzimas Cyp51 empleando la técnica in silico del acoplamiento molecular. Un incremento significativo de los valores de las energías libres de unión, con la consiguiente disminución de la constante de inhibición de los imidazoles 2-furilsustituidos con respecto a los fenilsustituidos, indicará una mayor afinidad de la proteína por los imidazoles 2-furilsustituidos. En caso contrario, indicará una mayor afinidad por los fenilsustituidos.
En la figura 2 se muestra cómo el sitio activo de esta proteína CYP51—L. infantum se encuentra en una región más bien neutra; se pudiera decir que hasta en algunos sitios ligeramente hidrofóbico, aunque la presencia del grupo Hemo le brinda un fuerte carácter hidrofílico. Por lo tanto, es de esperar que la mayor parte de las interacciones con los imidazoles ocurra mediante puentes de hidrógeno con el grupo Hemo e interacciones hidrofóbicas con el resto del sitio activo, aunque no se deben descartar otras interacciones.
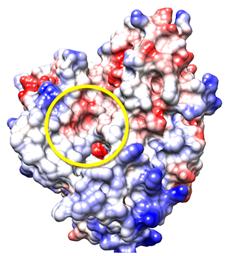
Fig. 2 Superficie electrostática de la proteína CYP51—L. infantum. Según la escala de colores, el rojo representa el carácter hidrofílico y el azul el hidrofóbico
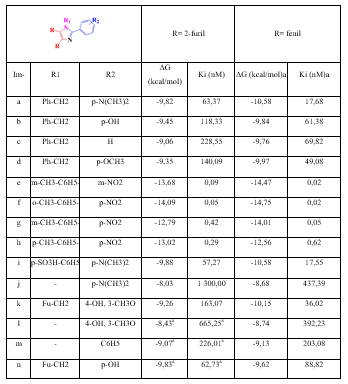
Los valores de las energías libres de unión y los valores de la constante de inhibición del complejo proteína-imidazol se muestran en la tabla 2. Como se puede observar, los imidazoles fenilsustituidos presentan mejores valores de energía libre de unión y de constante de inhibición que sus homólogos 2-furilsustituidos. Esto es algo esperado, ya que el grupo fenilo es más voluminoso que el 2-furilo y, por lo tanto, debe generar más interacciones hidrofóbicas. Sin embargo, un imidazol escapa a ese comportamiento.
Tabla 2 Valores de energías libres de unión (ΔG en kcal/mol) y constante de inhibición (Ki en nM) de los acoplamientos de los imidazoles sustituidos con la proteína CYP51—L. infantum
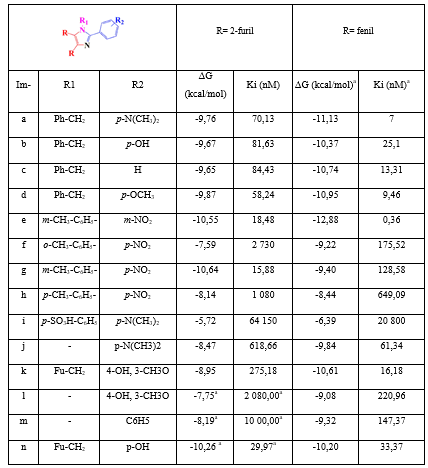
aFuente: R. VARGAS, J. A. et al.
El “Im g” presenta un descenso marcado en los valores de energía libre de unión y de constante de inhibición, por lo que se puede decir que es el único de los imidazoles 2-furilsustituidos con mejor afinidad hacia la proteína CYP51—L. infantum, que su par fenilsustituido. En la figura 3 se puede apreciar que las interacciones en común se establecen con los residuos Met357, Met359, Met459, Val460, Leu355, Tyr115, Phe104 y Tyr102. Aparte de ellas, “Im g” presenta 8 interacciones hidrofóbicas con residuos de aminoácidos y otra con el grupo Hemo. Por su parte, su par fenilsustituido presenta 6 interacciones hidrofóbicas con residuos de aminoácidos y tres puentes de hidrógeno con el grupo Hemo.
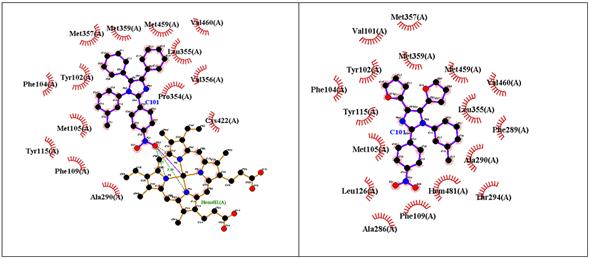
En la figura 4 se muestra cómo el sitio activo de esta proteína CYP51—T. brucei es similar al de la proteína CYP51—L. infantum, lo cual era de esperar, dado a que estos protozoos pertenecen a una misma familia.

Fig. 4 Superficie electrostática de la proteína CYP51—T. brucei. Según la escala de colores, el rojo representa el carácter hidrofílico y el azul el hidrofóbico
De manera similar ocurre con la proteína CYP51—T. brucei (tabla 3), donde los imidazoles fenilsustituidos presentan mejores valores de energía libre de unión y de constante de inhibición que sus homólogos 2-furilsustituidos, excepto para el imidazol “Im h”, el cual presenta un ligero descenso en su valor de energía libre de unión y constante de inhibición en comparación con su par fenilsustituido.
Tabla 3 Valores de energías libres de unión (ΔG en kcal/mol) y constante de inhibición (Ki en nM) de los acoplamientos de los imidazoles sustituidos contra la proteína CYP51— T. brucei
aFuente: R. VARGAS, J. A. et al.
En la figura 5 se puede apreciar que las interacciones en común de “Im h” y su par fenilsustituido se establecen con los residuos Met358, Met360, Met460, Val213, Cys422, Leu357, Tyr116, Phe105 y Tyr103. Aparte de ellas, “Im h” presenta 4 interacciones hidrofóbicas con residuos de aminoácidos y 3 puentes de hidrógeno con el grupo Hemo. Por su parte, su par fenilsustituido presenta 5 interacciones hidrofóbicas más con residuos de aminoácidos y también 3 puentes de hidrógeno con el grupo Hemo, aunque a mayor distancia que “Imi h”. Tal vez es por eso que la energía libre de unión de “Imi h” es mayor que la de su par fenilsustituido.
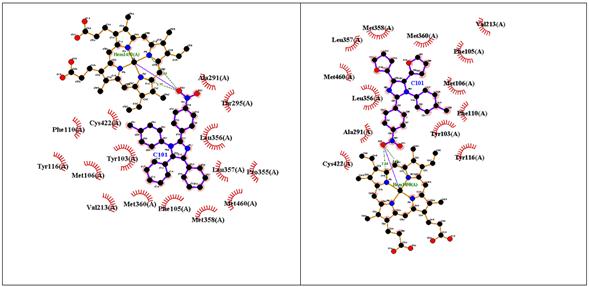
En la figura 6 se muestra cómo el sitio activo de esta proteína CYP51—T. cruzi es mucho más parecido al de la proteína CYP51—T. brucei que al de la proteína CYP51—L. infantum. De esto se podría esperar que el análisis de las conformaciones para esta proteína sea similar a la de la proteína CYP51—T. brucei y así es. En la tabla 4, se observar un ligero aumento de los valores de energía libre de unión y, por consiguiente menores valores de constante de inhibición para el imidazol “Im h”, al igual que ocurre en la proteína CYP51—T. brucei.
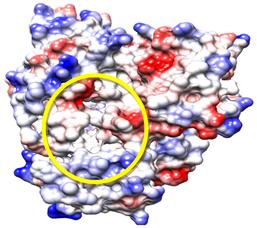
Fig. 6 Superficie electrostática de la proteína CYP51—T. cruzi. Según la escala de colores, el rojo representa el carácter hidrofílico y el azul el hidrofóbico
Tabla 4 Valores de energías libres de unión (ΔG en kcal/mol) y constante de inhibición (Ki en nM) de los acoplamientos de los imidazoles sustituidos contra la proteína CYP51— T. cruzi
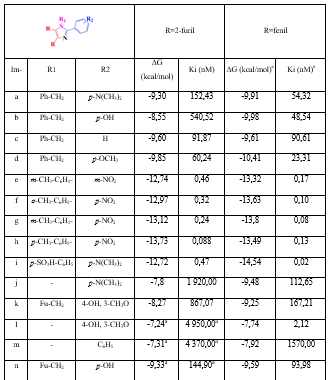
aFuente: R. VARGAS, J. A. et al.
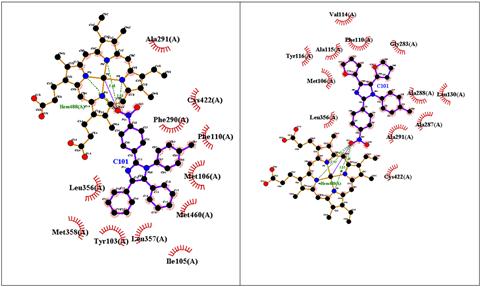
En la figura 7 se puede apreciar que las interacciones en común de “Im h” y su par fenilsustituido se establecen con los residuos Met106, Cys422, Leu 356, Ala291 y Phe110. Aparte de ellas, el “Im h” presenta 7 interacciones hidrofóbicas con residuos de aminoácidos y 5 puentes de hidrógeno con el grupo Hemo. Por su parte, su par fenilsustituido presenta 6 interacciones hidrofóbicas más con residuos de aminoácidos y 4 puentes de hidrógeno con el grupo Hemo, aunque a mayor distancia que “Imi h”.
Conclusiones
De manera general, los imidazoles sustituidos en las posiciones 4 y 5 con fenilos presentan mayor afinidad por las 3 proteínas estudiadas que los sustituidos con 2-furil. El “Imi g”, 4,5-di(furan-2-il)-2-(4-nitrofenil)-1-(m-toluil)-1H-imidazol constituye una excepción para la proteína CYP51— L. infantum, mientras que “Imi h”, 4,5-di(furan-2-il)-2-(4-nitrofenil)-1-(p-toluil)-1H-imidazol constiuye una excepción, tanto para la proteína CYP51— T. brucei como para T. cruzi.

