










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Ciencias Informáticas
versión On-line ISSN 2227-1899
Rev cuba cienc informat vol.10 no.3 La Habana jul.-set. 2016
ARTÍCULO ORIGINAL
Análisis de la Escalabilidad del cálculo paralelo de medidas de similitud entre pares de genes
Scalability Analysis of the parallel calculation of similarity measures between pair of genes
Addel Arnaldo Goya Jorge1*,Deborah Galpert Cañizares1, Reinier Millo Sánchez1, Claudia Companioni Brito1
1Universidad Central “Marta Abreu” de Las Villas, Cuba. Carretera Camajuaní km 5 1/2 Santa Clara. {agoya, rmillo, ccompanioni}@uclv.cu, deborah@uclv.edu.cu
*Autor para la correspondencia: agoya@uclv.cu
RESUMEN
El presente trabajo analiza la escalabilidad de una implementación paralela del cálculo de medidas de similitud entre pares en la detección de genes ortólogos. El análisis se realiza mediante el uso de métricas de calidad como la aceleración y la eficiencia que se calculan para algoritmos de cálculo del alineamiento par a par de secuencias y de cálculo de la similitud del perfil físico-químico de las proteínas. Los experimentos realizados en un conjunto de datos de dos genomas arrojan una mejora en el tiempo de ejecución de las implementaciones paralelas. No obstante, la escalabilidad de los algoritmos continúa siendo un objetivo de nuevas implementaciones.
Palabras clave: medidas de similitud, programación paralela, complejidad temporal, escalabilidad
ABSTRACT
This paper analyzes the scalability of the parallel implementation of pairwise similarity measures for gene comparison in ortholog gene detection. This analysis is carried out by using metrics such as speedup and efficiency that are calculated for all vs all alignment and physicochemical profile comparisons. The experiments with a dataset of two genomes show an improvement in the execution time of the parallel implementations. However, scalability continues to be a goal for further implementations.
Key words: similarity measures, parallel computing, time complexity, scalability.
INTRODUCCIÓN
La comparación de genes es imprescindible en problemas de la Bioinformática como la comparación de genomas, donde se detectan las similitudes entre los genomas para dar respuesta a interrogantes como la función de proteínas desconocidas que se encuentran en regiones conservadas (similares) (Kamvysselis, 2003). Aquellas proteínas producidas por genes que generalmente conservan su función son reconocidas mediante la detección de genes llamados ortólogos entre dos o más genomas. Estos son los genes que se transfieren de un genoma a otro en la evolución mediante un evento de especiación. Son considerados genes homólogos porque tienen un gen ancestro común en las especies que se estudian (Fitch, 1970). En la detección de los ortólogos, estos deben distinguirse de otro tipo de genes homólogos llamados parálogos que se producen por un evento de duplicación en un mismo genoma (Kuzniar et al., 2008).
Para detectar los genes homólogos, y en específico los ortólogos, se han utilizado métodos de alineamiento de secuencias como el BLAST (Altschul et al., 1997) cuyas puntuaciones se utilizan para comparar el grado de similitud entre las secuencias. Sin embargo, sucede que los algoritmos de detección basados únicamente en dicha similitud han sido fuertemente criticados porque producen falsos positivos ante la presencia de parálogos y pérdidas de genes (Kuzniar et al., 2008), (Salichos and Rokas, 2011). Además producen falsos negativos cuando las secuencias son cortas o divergentes (alejadas en la evolución) (Kuzniar et al., 2008). Para complementar la información de similitud de las secuencias se han propuesto otras informaciones relacionadas con la vecindad conservada de los genes (Kristensen et al., 2011), (Lechner et al., 2014).
Con el fin de combinar diversas informaciones en la clasificación de genes, en (Millo et al., 2014) se han presentado medidas de similitud para comparar pares de genes y métodos de clasificación que agregan estas medidas en algoritmos no supervisados o las combinan en modelos supervisados (Galpert et al., 2014), (Galpert et al., 2015) . El enfoque de combinación de medidas de similitud abarca dos fases: (i) cálculo de medidas de similitud para pares de genes y (ii) clasificación por pares. De acuerdo al total de posibles pares de genes de un par de genomas, así como al continuo crecimiento del total de genomas anotados, en espera de ser comparados para su estudio (Sonnhammer et al., 2014), se hace necesario, en primera instancia, estudiar la escalabilidad de los algoritmos de cálculo de las medidas de similitud y aplicar técnicas de programación paralela a este problema, una vez que se analice la ventaja del uso de estas técnicas en cuanto a su desempeño en tiempo de ejecución. Para esto, en este trabajo se analiza el rendimiento de los algoritmos de cálculo de dichas medidas de similitud para pares de genes mediante métricas de desempeño de implementaciones paralelas como la aceleración y la eficiencia.
Análisis de la escalabilidad de algoritmos secuenciales y paralelos
La escalabilidad es la habilidad de un sistema o proceso de manejar un aumento creciente en el volumen de trabajo a realizar de una manera competente, o su habilidad de ser ampliado para acomodar este crecimiento. Se puede referir a la capacidad de un sistema de aumentar su salida total bajo una carga aumentada cuando se añaden recursos (típicamente hardware) (Bonvin, 2012). Para analizar la escalabilidad de algoritmos paralelos en cuanto al tiempo de ejecución se analiza su rendimiento en condiciones de aumento de la carga utilizando métricas como la aceleración y la eficiencia que se especificarán más adelante en esta sección.
El rendimiento de algoritmos secuenciales se evalúa por su tiempo de ejecución en función de la dimensión del problema, es decir, en función del tamaño de los datos de entrada. Para este análisis comúnmente se utiliza la notación asintótica que permite acotar el tiempo de ejecución de un algoritmo y así especificar su orden de complejidad temporal (Brassard, 1997). Sin embargo, para analizar la complejidad temporal de los algoritmos paralelos se deben tener en cuenta varios factores como son: el tamaño del problema, el número de procesadores y determinados parámetros de comunicación de la plataforma sobre la que se ejecuta el algoritmo (Grama et al., 2003).
En (Grama et al., 2003) se plantea que el rendimiento de un algoritmo paralelo puede ser analizado con una mayor exactitud cuando se compara con su mejor versión secuencial, o en última instancia con alguna versión secuencial del mismo. En base al tiempo de ejecución de la versión secuencial Ts y el tiempo de la versión paralela Tp se calculan métricas como la aceleración y la eficiencia.
La aceleración ![]() es una medida que captura el beneficio relativo de resolver un problema en paralelo. Se define como el radio de tiempo que se toma para resolver un problema en un único elemento de procesamiento con relación al tiempo requerido para resolver el mismo problema con P procesadores. Sólo un sistema ideal puede lograr una aceleración igual a P. Para ilustrar la aceleración real, es posible utilizar constantes en lugar de la notación asintótica (Grama et al., 2003) como se verá en la siguiente sección.
es una medida que captura el beneficio relativo de resolver un problema en paralelo. Se define como el radio de tiempo que se toma para resolver un problema en un único elemento de procesamiento con relación al tiempo requerido para resolver el mismo problema con P procesadores. Sólo un sistema ideal puede lograr una aceleración igual a P. Para ilustrar la aceleración real, es posible utilizar constantes en lugar de la notación asintótica (Grama et al., 2003) como se verá en la siguiente sección.
La eficiencia, por otra parte, es una medida de la fracción de tiempo para la cual un elemento de procesamiento es útilmente empleado. Se define como ![]() En un sistema ideal la eficiencia es 1. En la práctica la aceleración es menor que P y la eficiencia está entre cero y uno. La eficiencia de los programas paralelos decrece con el aumento del número de elementos procesamiento para un tamaño de problema dado (Grama et al., 2003).
En un sistema ideal la eficiencia es 1. En la práctica la aceleración es menor que P y la eficiencia está entre cero y uno. La eficiencia de los programas paralelos decrece con el aumento del número de elementos procesamiento para un tamaño de problema dado (Grama et al., 2003).
Escalabilidad del cálculo paralelo de medidas de similitud para pares de genes
En esta sección se especifican, en código MATLAB (2010), los procedimientos de cálculo de dos medidas de similitud, definidas en (Millo et al., 2014), para comparar una secuencia de un genoma con todas las secuencias de otro genoma. Estos procedimientos tienen la mayor complejidad temporal con relación a los de otras medidas por lo que se propone su paralelización. El primero se corresponde con el cálculo del alineamiento de una secuencia de un genoma con todas las secuencias de otro, usando el algoritmo secuencial de la Figura 1 o el paralelo de la Figura 2, y el segundo, con el cálculo de la similitud basada en el perfil físico-químico de todas las proteínas de ambos genomas usando el algoritmo secuencial de la Figura 3 o el paralelo de la Figura 4.



Las funciones swaling y nwalign del MATLAB tienen una complejidad computacional de O(m × n), donde m y n son las longitudes máximas de las secuencias de cada genoma, y como la longitud máxima que puede tener el alineamiento de las secuencias es m + n entonces la función calculate_similarity tiene una complejidad de O(m+n), siendo entonces la complejidad computacional de la versión secuencial de la similitud basada en el alineamiento O(N × m × n), donde N es el total de ciclos a realizar. Los tiempos secuencial y paralelo correspondientes al cálculo de los alineamientos son referenciados como Ts1 y Tp1.
La función calculate_profile_feature tiene una complejidad de O((m + n)2), siendo la complejidad computacional de la versión secuencial del cálculo del perfil físico-químico O(N × (m+n)2). En este caso, los tiempos secuencial y paralelo correspondientes a este cálculo serán referenciados como Ts2 y Tp2 en las ecuaciones (1) y (2), respectivamente.
Teniendo en cuenta la complejidad computacional y la estructura de los algoritmos secuenciales, los tiempos de ejecución secuenciales se pueden estimar como:

donde tc indica el tiempo requerido para realizar las operaciones aritméticas. Para el desarrollo de este trabajo asumiremos el valor de tc óptimo, tc = 1. El tiempo de ejecución paralelo TP se define en función del tiempo de cálculo y el tiempo de comunicación entre los procesadores. En el caso de la versión paralela el tiempo de cálculo viene dado por la distribución de los N ciclos entre los P procesadores empleados. Con la distribución de los ciclos, pueden darse los casos en que: todos los procesadores realizan [N/P] iteraciones, o p < P procesadores realizan [N/P]+1 iteraciones; por lo que tomando el peor de los casos, el tiempo de cálculo de cada una de las versiones paralelas se puede estimar como aparece en las ecuaciones (3) y (4):

El tiempo de comunicación entre los procesadores está directamente relacionado con la información accedida en la sección secuencial del algoritmo. En los dos casos que se analizan, en cada iteración cada uno de los P procesadores necesita recibir la información de las secuencias que se alinean. Siendo m y n las longitudes de las dos secuencias, el tiempo de comunicación se expresa como aparece en la ecuación (5):
![]()
donde ts representa el tiempo necesario para establecer la comunicación y preparar la información a enviar; y tw el tiempo necesario para enviar un valor numérico. Para el desarrollo de este trabajo asumiremos estos valores como óptimos, ts= 0 y tw= 1. De esta forma, al combinar la información de las ecuaciones (3) y (5) se obtiene una estimación del tiempo de ejecución paralelo para el cálculo del alineamiento (ecuación (6)) y combinando las ecuaciones (4) y (5) se obtiene una estimación del tiempo de ejecución paralelo para el cálculo del perfil (ecuación (7)).

Empleando las expresiones TS y TP para cada uno de los algoritmos, se realizó un análisis del comportamiento de la aceleración y la eficiencia para diferentes muestras de N, variando la cantidad de procesadores entre 1 y 6000. En las Figuras 5,6,7,8 se muestran las gráficas de los resultados obtenidos para un conjunto de datos de comparación de los genomas de Sccharomyces Scerevisiae y Schizosaccharomyces Pombe. Para el caso del alineamiento, el 100% de la muestra está dado por las 5006 secuencias del genoma del S. Pombe que se comparan con una secuencia de S. Scerevisiae, y para el perfil, por 16324500 pares de secuencias. Se puede observar que al mantener constante el tamaño de la muestra y aumentar la cantidad de procesadores, el valor de la aceleración y la eficiencia tienden a disminuir. No siendo así cuando se mantiene constante la cantidad de procesadores y se aumenta la muestra. Cuando la cantidad de procesadores alcanza el tamaño de la muestra, la aceleración y la eficiencia comienzan a decrecer hasta que alcance un valor constante.



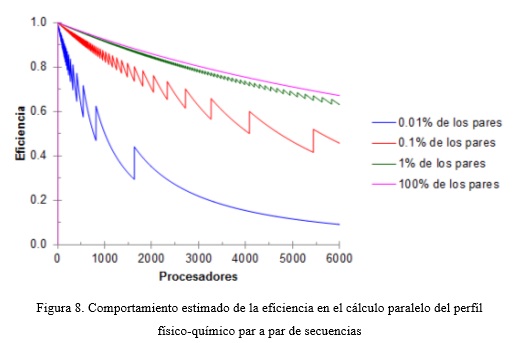
En ocasiones en que es casi imposible la ejecución secuencial del algoritmo, estos valores estimados pueden dar un valor cercano a la realidad. El cálculo de las medidas de similitud empleando ambos algoritmos paralelos fue realizado usando 6 procesadores, por lo que el algoritmo paralelo para el alineamiento se ejecutó aproximadamente 5.98 veces más rápido que la versión secuencial, con una eficiencia de 0.9968; y el algoritmo paralelo para el perfil se ejecutó aproximadamente 5.9971 veces más rápido que la versión secuencial, con una eficiencia de 0.9995.
A partir de los tiempos de ejecución en paralelo de las ecuaciones (6) y (7), se pueden estimar las complejidades computaciones de cada uno de los algoritmos cuando su ejecución es en paralelo. En ambos casos, la expresión ([N/P]+1) se puede tomar como [N/P]. En el caso del algoritmo para el cálculo de la similitud basada en el alineamiento, los elementos 2 × m × n y (m + n) × (P + 1) no se pueden comparar debido a la presencia de P. Omitiendo los valores de las constantes, su complejidad temporal está dada como ![]() , la cual es menor que O(N × m × n), como se muestra en la ecuación (8). Por otra parte, en el algoritmo del cálculo del perfil se tiene que m × n £ (m + n) × (m + n + P), por lo que su complejidad temporal es,
, la cual es menor que O(N × m × n), como se muestra en la ecuación (8). Por otra parte, en el algoritmo del cálculo del perfil se tiene que m × n £ (m + n) × (m + n + P), por lo que su complejidad temporal es, ![]() la cual es menor que O(N ×(m+n)2), como se muestra en la ecuación (9).
la cual es menor que O(N ×(m+n)2), como se muestra en la ecuación (9).

Usando las versiones secuenciales y paralelas de cada uno de los algoritmos, se calculó el tiempo de ejecución secuencial y los tiempos de ejecución en paralelo usando 1, 2, 3 y 4 procesadores. Para esto se utilizó una computadora con procesador Intel ® CoreTM i3 CPU M380 a 2.53 GHz, con una memoria RAM DDR3 de 4,0 Gb, con MATLAB(R2010a)(2010)sobre el sistema operativo Windows 7 de 64Bits. Los experimentos fueron realizados sobre el 50% de los pares y el conjunto total de los pares. Para el caso del cálculo del perfil se realizó una selección aleatoria de 1921 pares que representan el 100%. Las Figuras 9, 10, 11, 12 muestran los resultados obtenidos. Como se observa en estas figuras los valores estimados tienen un comportamiento similar entre sí, al igual que los valores reales calculados a partir de los tiempos de ejecución. Los mejores valores de aceleración se obtienen para la ejecución sobre 4 procesadores, pero con una mayor eficiencia cuando la ejecución fue sobre 2 procesadores.
Teóricamente el valor de la aceleración se encuentra acotado por la cantidad de procesadores empleados en la ejecución, y la eficiencia oscila en el intervalo [0, 1]. Sin embargo, en este caso la aceleración es superior a la cantidad de procesadores empleados en cada ejecución y el valor de eficiencia es superior a 1. A este fenómeno se le conoce como aceleración super-lineal, es poco común, y se produce porque cada elemento de procesamiento consume un tiempo inferior a la razón TS/P. Esto puede estar debido a que la versión paralela realiza menos trabajo que la versión secuencial, o a factores relacionados con los recursos que se emplean, como por ejemplo, la memoria caché (Grama et al., 2003).




CONCLUSIONES
Con el uso de la instrucción parfor para el cálculo en paralelo de los alineamientos par a par de secuencias y el perfil físico-químico de las proteínas, se logra reducir la complejidad computacional del cálculo de estas medidas.
No obstante, la habilidad del sistema de mantener la eficiencia en un valor fijo al incrementar simultáneamente el número de procesadores y el tamaño del problema se mantiene como objetivo de futuras implementaciones. Con esta implementación no se puede decir que el sistema es escalable (Grama et al., 2003) en todo su espectro, por lo que se pretende en próximos trabajos modificar la implementación en función de mejorar la escalabilidad, necesaria para lograr las clasificaciones de genes.
REFERENCIAS BIBLIOGRÁFICAS
2010. Matlab [Online]. Available: http://www.mathworks.com Accessed 2012.
BONVIN, N. 2012. Linear Scalability of Distributed Applications. ÉCOLE POLYTECHNIQUE FÉDÉRALE DE LAUSANNE.
BRASSARD, G., BRATLEY, T. (ed.) 1997. Fundamentos de Algoritmia, Madrid: Prentice Hall.
GALPERT, D., RÍO, S. D., HERRERA, F., ANCEDE-GALLARDO, E., ANTUNES, A. & AGÜERO-CHAPIN, G. 2015. An Effective Big Data Supervised Imbalanced Classification Approach for Ortholog Detection in Related Yeast Species. BioMed Research International [Online], 2015, Article ID 748681.
LECHNER, M., HERNANDEZ-ROSALES, M., DOERR, D., WIESEKE, N., THÉVENIN, A., STOYE, J., HARTMANN, R. K., PROHASKA, S. J. & STADLER, P. F. 2014. Orthology Detection Combining Clustering and Synteny for Very Large Datasets. PLoS ONE, 9(8), e105015.
SONNHAMMER, E. L. L., GABALDÓN, T., SILVA, A. W. S. D., MARTIN, M., ROBINSON-RECHAVI, M., BOECKMANN, B., THOMAS, P. D. & DESSIMOZ, C. 2014. Big data and other challenges in the quest for orthologs. Bioinformatics Editorial, 1-6.
Recibido: 24/02/2016
Aceptado: 04/07/2016

