










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Farmacia
Print version ISSN 0034-7515
Rev Cubana Farm vol.46 no.1 Ciudad de la Habana Jan.-Mar. 2012
ARTÍCULO ORIGINAL
Cuantificación de glibenclamida en muestras de limpieza de equipos farmacéuticos mediante cromatografía líquida de alta resolución
Quantification of glibenclamide in cleaning samples of pharmaceutical equipment through high performance liquid chromatography
MSc. Alen Nils Baeza Fonte,I Lic. Elsa Eneida Díaz Águila,II Dra. C. Nancy Martínez AlfonsoIII
I Instituto de Ciencia y Tecnología de Materiales. Laboratorio de Análisis Químicos (LAQ). Universidad de La Habana. La Habana, Cuba.
II Laboratorios MedSol. La Habana, Cuba.
III Facultad de Química. Universidad de La Habana. La Habana, Cuba.
RESUMEN
Objetivo: proponer un procedimiento analítico selectivo para la cuantificación de glibenclamida en muestras de limpieza de equipos farmacéuticos mediante cromatografía líquida de alta resolución.
Métodos: la fase móvil consistió en una mezcla equivalente de volúmenes de acetonitrilo y solución amortiguadora KH2PO4 de concentración 0,037 mol/L a pH 5,25 y flujo 1,5 mL/min, en una columna Nucleosil 100 C8. La glibenclamida se inyectó con progesterona como estándar interno y empleando detector UV a una l= 230 nm.
Resultados: el método resultó lineal en el intervalo de concentraciones de 0,4-150 mg/mL, teniendo como límites de detección y cuantificación 10 y 40 ng/mL respectivamente y siendo específico al analito en presencia del placebo, sus productos de degradación y a otros ingredientes farmacéuticamente activos. Se consideraron potenciales de interferencias para el método propuesto: captopril, clortalidona, dexametasona, digoxina, 8-cloroteofilina, difenhidramina HCl, fenobarbital, haloperidol, hidroclorotiazida, ácido fumárico, ketotifeno, metoclopramida HCl, piridoxina HCl, piroxicam, prednisona y nifedipino. Se identificaron: ibuprofeno, indometacina, trifluoperazina HCl, tioridazina HCl e imipramina HCl, como interferentes del procedimiento en concentraciones cercanas a 10 mg/mL.
Conclusiones: el método desarrollado es sensible, rápido y especialmente selectivo para la evaluación de residuales del principio activo glibenclamida en equipos de producción de tabletas, empleando un muestreo por hisopado, y pudiera utilizarse potencialmente cuando exista sospecha de contaminación cruzada de glibenclamida con otros fármacos de los aquí descritos.
Palabras clave: glibencamida, gliburida, CLAR, cromatografía líquida de alta resolución, validación, validación de limpieza, trazas, límite de detección, límite de cuantificación.
ABSTRACT
Objective: to submit a selective analytical method for quantization of glibenclamide in cleaning samples of pharmaceutical equipment using high performance liquid chromatography.
Methods: the mobile phase consisted of an equal mixing of acetonitrile/phosphate buffer KH2PO4; with 0.037 mol/L concentration pH 5.25 and flow of 1.5 mL/min, in a Nucleosil 100 C8 column. Glibenclamide was injected with progesterone as internal standard and using an UV detector= 230 nm
Results: the method was linear in the 0.4-150 mg/mL concentration interval having a detection and quantization limits of 10 and 40 ng/mL respectively. It was specific to analyte when placebo is present, to degradation products and to other active ingredients. Possible interferences with the proposed method was considered for captopril, chlortalidone, dexametasone, diphenhydramin HCl, digoxine, 8-chlortheophylline, diphenhydramina HCl, phenobarbital, haloperidol, hydrochlorothiazide, fumaric acid, ketotifen, metoclopramide HCl, piridoxine HCl, piroxicam, prednisone and nifedipine, On the other hand, ibuprofen, indometacin, trifluoperazine HCl, thioridazine HCl and imipramine were identified as interferences in the procedure at concentration figures close to 10 mg/mL.
Conclusions: the present method is sensitive, quick and selective for the evaluation of residues of active pharmaceutical principle glibenclamide in tablet production equipment after a swap sampling and it could be potentially used in case of cross-contamination of glibenclamide and other drugs already described.
Key words: glibenclamide, glyburide, high performance liquid chromatography, validation, cleaning validation, traces, detection limit, quantization limit.
INTRODUCCIÓN
El cumplimiento de las Buenas Prácticas de Fabricación en la Industria Farmacéutica es un paso vital para asegurar la calidad de cualquier medicamento, más aun en las facilidades multiproducto donde hay que minimizar la contaminación cruzada entre aquellos que se elaboran en un mismo proceso o equipo. Tomando en cuenta esta situación se establecen procedimientos de limpieza, cuya verificación se realiza mediante el análisis químico.
El mayor problema en esta industria es la presencia de múltiples impurezas que aparecen durante el muestreo, las que se relacionan con residuos de otros ingredientes farmacéuticamente activos (IFA) cuya elaboración se realiza en los mismos equipos1 o excipientes acompañantes, lo que imposibilita la aplicación directa de métodos existentes por la carencia de especificidad y por el tiempo de duración del análisis, ya que el equipo en evaluación queda en cuarentena mientras se realiza el procedimiento.
Los métodos usualmente empleados en validaciones de limpieza, como es el caso, se clasifican en dos grupos: los específicos y los que no lo son. En el primer grupo se ubican la cromatografía tanto líquida (CLAR) como gaseosa, espectrometría de absorción atómica y la electroforesis capilar; la CLAR es la más empleada por su versatilidad, sensibilidad , selectividad y amplio uso en el análisis de sulfonilureas como glibenclamida (Glib).2-5 En el segundo grupo se encuentran el análisis de carbono orgánico total, medidas de pH, conductimetría y determinación de sólidos totales entre otros.6,7
Para determinar el criterio de limpieza existen normativas que establecen factores de seguridad a partir de la dosis, forma farmacéutica, toxicidad y vía de administración. Otros criterios se basan en que estos valores de residuos en ningún caso deben sobrepasar el 0,1 % de la dosis diaria mínima o no más de 10 mg/kg de cualquier residuo en otro medicamento que se fabrique posteriormente al analizado, y de esta manera, establecer así los casos más críticos que definirán el límite de limpieza.8-11
El objetivo de este trabajo es proponer un método por CLAR para la evaluación de trazas de Glib en muestras de limpieza.
MÉTODOS
Reactivos: fosfato de potasio monobásico (KH2PO4, p.a. 99,5 %); hidróxido de sodio 0,1 mol/L; ácido clorhídrico 0,1 mol/L; acetonitrilo (chromasolv, 99,8 %); metanol (chromasolv, 99,8 %); todos de la firma Riedel-de Haën; sulfanilamida (p.a., 99,0 %, Fluka) y agua bidestilada (Bidestilador GFL-2104).
Equipos: cromatógrafo KNAÜER conformado por una bomba K-1001, desgasificador y organizador de solvente K1500 de la serie WellChrom, detector UV-Visible Smartline 2500 e inyector REODYNE 7725i, balanza analítica Sartorius, precisión 10-5 g, espectrofotómetro Jasco V-530 y pHmetro Radiometer PHM-82.
Materias primas: Glib, materia prima (Effechem, Italia), placebos correspondientes a las tabletas de glibenclamida elaborados según técnica del fabricante; clortalidona, materia prima (IFF SPA, Italia).
Materiales de referencia: Glib, dexametasona, difenhidramina HCl, ibuprofeno, propranolol HCl, dimenhidrinato, fenobarbital, haloperidol, hidroclorotiazida, imipramina HCl, indometacina, ketotifeno fumarato, metoclopramida HCl, piridoxina HCl, piroxicam, prednisona, tioridazina HCl, trifluoperazina HCl, nifedipino, ketoconazol, prednisolona, todos estándares de referencia certificados (CIDEM, Subdirección de Control Técnico encargada del Suministro Nacional de Patrones a la Industria Farmacéutica Cubana) y digoxina (USP).
Métodos computacionales y softwares: programa de cómputo ULC (Univariate Linear Calibration, departamento de química analítica de la universidad de tarragona); Excel, Statgraphics Plus versión 5.1 y Eurochrom 2000.
Establecimiento del límite de limpieza
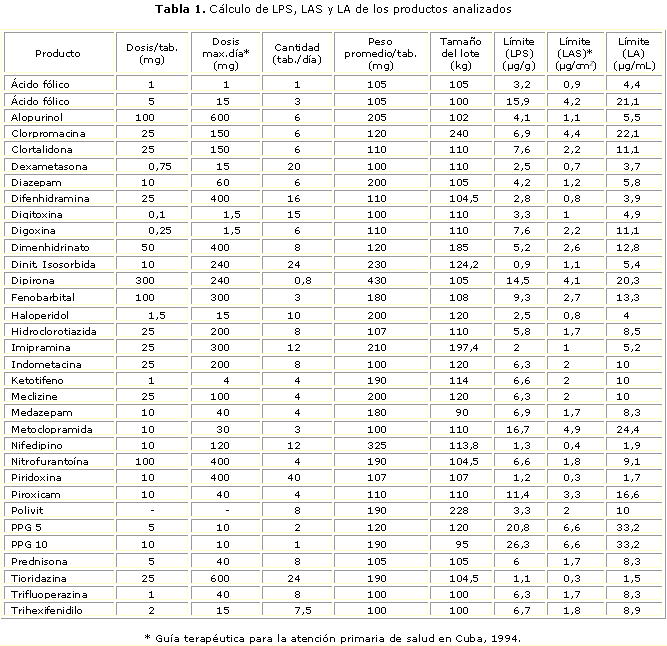
Los criterios más recientes plantean que los límites deben ser calculados según la mínima dosis diaria del producto activo a limpiar9 (producto A) en la máxima dosis del producto siguiente (producto B) (ecuación 1), empleando un factor de seguridad de 0,001 (medicamentos orales). El valor obtenido de límite en el producto siguiente (LPS) se compara con 10 mg/kg, escogiendo el menor de ambos.
LPS= 0,001 × mín. dosis activa diaria de A / máx. dosis diaria de B (1)
El límite por área de superficie (LAS) se define cuando se expresa el LPS en términos del nivel de contaminación del PA por área de superficie del equipo (LAS), expresado en microgramo por centímetro cuadrado (mg/cm2) y depende del LPS escogido (valor menor entre LPS y 10 mg/kg), del tamaño del lote del producto B (TL) y del área total del equipamiento en contacto con ambos productos A y B (AT), expresado en centímetro cuadrado (cm2) (ecuación 2).
LAS= LPS × TL / AT *1000 (2)
Para las facilidades multiproducto, debe calcularse el LAS del producto a limpiar con cada producto que se elabore y escoger el menor valor de los obtenidos.
En el caso del muestreo, es necesario conocer el área muestreada (AM), donde en general se escogen 25 cm2 en cada punto, y el volumen en que se desorbe la muestra tomada por hisopado (VD); de esta forma, el límite analítico (LA), se calcula según la ecuación 3:
LA= LAS × AM / VD (3)
Es necesario aclarar que durante el desarrollo del método no se realizó hisopado, solo se tomó en cuenta para determinar la concentración final de la muestra de referencia durante los estudios considerando un VD de 5 mL.
Selección de la fase móvil
Para la evaluación de las fases móviles se calculó el parámetro de solubilidad (dm, ecuación 4), asumiendo que para el agua su valor es aproximadamente cero en fase invertida, representada por la solución amortiguadora, siendo la del MeOH y el del ACN 2,6 y 3,2 respectivamente:12
d m= S ji C di (4)
donde :
ji: la fracción volumétrica del solvente en la FM
di: parámetro de solubilidad de cada componente en la FM .
Considerando que las mezclas binarias cumplen que:
dACN/KH2PO4 0,037 mol/L= jACN ´ dACN + jBuffer ´ dBuffer (5)
dMeOH/KH2PO4 0,037 mol/L= jMeOH ´ dMeOH + jBuffer ´ dBuffer (6)
Siendo d (buffer) igual a cero se tiene que:
dACN/ KH2PO4 0,037 mol/L= jACN ´ dACN (7) y
dMeOH/ KH2PO4 0,037 mol/L= jMeOH ´ dMeOH (8)
A partir de las expresiones 7 y 8 se prepararon cuatro FMs a pH 5,25 y 3,25 cada una con las siguientes composiciones solvente orgánico/KH2PO4 0,037 mol/L (V/V):
a) ACN 50 %, jACN= 0,5; d=1,6
b) MeOH 50 %, jMeOH= 0,5; d=1,3
c) MeOH 62 %, jMeOH= 0,62; d=1,6
d) ACN 41 %, jACN= 0,41 %; d=1,6
Se ajustan cada una a los pH 5,25 y 3,25; se inyectan 500 mL de disolución de Glib (1,50 mg/mL) en la columna Nucleosil C8 a 1 mL/min; se disuelven con MeOH/agua 70/30 o ACN/agua 60/40 y se detectan a 225 nm y a 230 nm respectivamente en correspondencia con el componente orgánico de la FM a 25 ± 2 °C, en régimen isocrático. Ambas lambdas fueron determinadas previamente a partir de un espectro de absorción entre 200 y 350 nm en cada FM. Se empleó como estándar interno progesterona (Prog) a una concentración de 1,80 mg/mL.
Especificidad los productos de degradación y placebo: se preparó una solución con los productos de degradación de Glib: metil-N-4-[2-(5-cloro-2-metoxibenzamido)etil]bencenosulfonilcarbamato (carbamato) y 5-cloro-2-metoxi-n-[2-(4-sulfamoifenil)etil]benzamida (bencenosulfonamida) en las concentraciones de 0,30 mg/mL para ambos productos; 0,75 µg/mL para Glib; 1,80 mg/mL para Prog; 28,0 mg del placebo y 30 mL de FM. Después de agitar durante 1 h fue determinado el grado de interferencia de estos con respecto a Glib y Prog.
Parámetros de validación
Linealidad: la curva de calibración fue construida a partir de soluciones de concentración 0,40; 0,80; 1,20; 2,00; 4,00 y 10,00 y 20,00 mg/mL por triplicado cada una con Prog en una concentración de 1,80 mg/mL empleando un lazo de 500 mL, con vistas a aumentar la sensibilidad del método y cumplir con los criterios acordes a la literatura.10,11 Para el análisis de los resultados se graficó la relación de áreas de Glib/Prog vs. concentración, calculando los coeficientes de regresión (r), determinación (r2), pendiente, intercepto y significación de estos.
Límites de detección (LD) y cuantificación (LC): se determinaron a partir de tres soluciones de concentración 10, 40 y 100 ng/mL, cercanas al LC del método analítico, calculando la relación señal/ruido del analito con respecto a la línea base.
Precisión: se evaluó a través de la repetibilidad y la precisión intermedia. Para la primera fueron considerados tres niveles de concentración 0,75; 1,50 y 15,0 mg/mL por sextuplicado que representaron el 50, 100 y 1 000 % con respecto al valor máximo permisible para el LA a una concentración de 1,50 mg/mL, determinando la influencia de este factor mediante la prueba de Cochran y el coeficiente de variación (CV) para cada uno. Para la segunda fue tomada en cuenta una sola concentración correspondiente al LA por cuatro analistas en 3 d distintos.
Exactitud: se analizó por el método de los placebos cargados trabajando con soluciones de concentración 0,75; 1,12; 1,50; 1,88; y 2,25 mg/mL de Glib que corresponden con 50, 75, 100, 125 y 150 % de la concentración estimada como LA, previa adición de 28,0 mg de placebo.
RESULTADOS
En este estudio a partir del cálculo de LPS y LA se determinó que la Glib, en su forma terminada tableta 5 mg, fabricada mediante proceso completo, mostró el nivel de contaminación permisible más bajo entre los calculados (tabla 1); por tanto, fue el IFA que fijó el método de limpieza y los niveles a alcanzar, cuantificado según CLAR, asumiendo un muestreo por hisopado. El LA se estimó en 1,50 mg/mL equivalente a un LAS de 7,5 mg para un área de muestreo de 25 cm2, el cual permitió establecer los criterios de aplicabilidad del método.
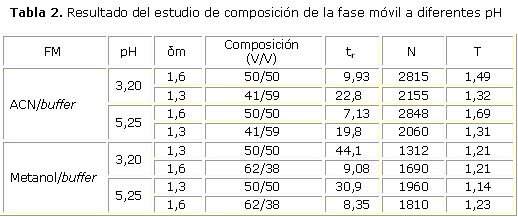
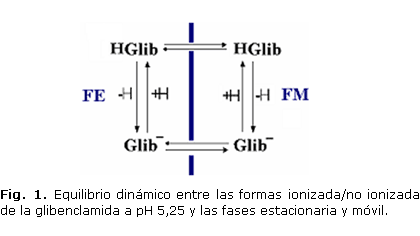
El desempeño de la FM y pH de trabajo fue determinado a partir de los parámetros cromatográficos tr, N y T; las condiciones más idóneas resultaron las relacionadas con la FM ACN/KH2PO4 0,037 mol/L, 50/50 (V/V) a pH 5,25 midiendo a 230 nm (tabla 2), tomando en consideración el equilibrio que manifiesta la Glib en disolución durante la cromatografía (Fig. 1). Durante la optimización de los flujos se realizaron variaciones de este en 0,25 unidades entre 1 y 2 mL, tomando como valor de trabajo 1,50 mL/min por mostrar valores de presión relativamente bajos (8-10 atm) y menores tr con respecto al flujo inicial de 1 mL/min, sin perder resolución con respecto al resto de los analitos.
Especificidad
El cromatograma obtenido con productos de degradación de la Glib, según USP 30, no mostró ninguna interferencia. Los tr obtenidos fueron: bencenosulfonamida (2,95), carbamato (3,49), Glib (5,82), Prog (8,09) y una señal procedente del algodón del hisopo que precedió a todos ellos (Fig. 2). Los excipientes acompañantes, que conformaron el placebo, no mostraron señal analítica en el tiempo de la corrida.

Los IFA identificados como posibles interferencias fueron cromatografiados determinando su grado de afectación en la cuantificación de Glib (tabla 3).
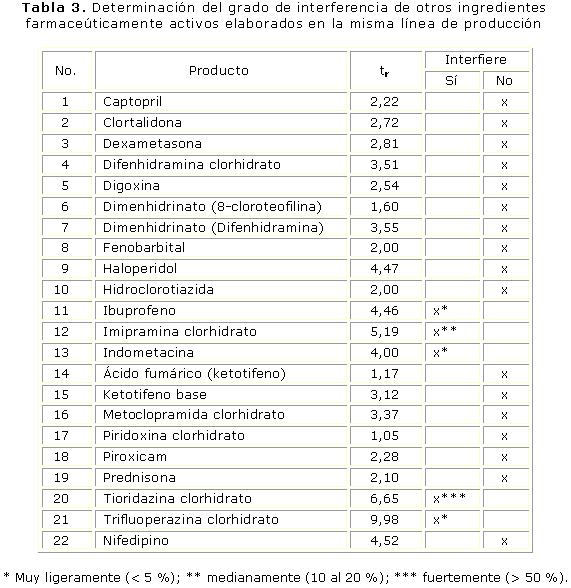
La especificidad a otros posibles contaminantes fue realizada sobre disoluciones independientes de concentración 10 µg/mL en FM de los IFA captopril, clortalidona, dexametasona, difenhidramina HCl, digoxina, 8-cloroteofilina, difenhidramina HCl, fenobarbital, haloperidol, hidroclorotiazida, ketotifeno fumarato, metoclopramida HCl, piridoxina HCl, piroxicam, prednisona, nifedipino, ibuprofeno, indometacina, trifluoperazina HCl, tioridazina HCl e imipramina HCl inyectadas bajo las mismas condiciones cromatográficas.
Los estudios de linealidad mostraron resultados favorables de r= 0,9999; r2= 0,9999 y los p-valores asociados al intercepto y a la pendiente tuvieron valores de 0,21 y 0,00. Durante el estudio del LD y LCD se inyectaron las soluciones correspondientes a los niveles de concentración de 10, 40 y 100 ng/mL que produjeron CV de 5,80; 3,90 y 5,66 %; mientras que la relación señal/ruido de las dos primeras tuvo valores de 5,1 y 12,5.
En la repetibilidad se obtuvieron CV de 1,40; 0,87 y 0,64 % para las concentraciones de 0,75; 1,50 y 15 mg/mL respectivamente. La prueba de Cochran mostró un p-valor de 0,51.
El análisis entre días, durante la precisión intermedia, mostró un p-valor para la prueba de Cochran de 0,23; mientras que entre analistas su valor fue de 0,50. El CV total obtenido para el ensayo tuvo un resultado de 1,97 %.
En la exactitud no se observaron desviaciones del recobrado con respecto a los valores teóricos, con resultados experimentales entre 98,8-101,4 %, mientras que la prueba de Cochran tuvo un p-valor de 0,37.
La estabilidad de la solución estándar de Glib 1,50 mg/mL se mantuvo por 72 h a temperatura ambiente sin que aparecieran productos de degradación manteniéndose la relación de áreas Glib/Prog con un CV de 1,21 % entre los intervalos de muestreos.
DISCUSIÓN
La FM escogida proporciona menores tr en comparación con las otras, parámetro crítico, por cuanto la espera del resultado analítico demora la producción incrementando los costos.
Los valores de T son inferiores a 2,0, lo cual indica una elevada simetría del pico y además mayor N lo que implica un equilibrio de interacción del analito mucho más rápido.
La Glib, con un pKa de 5,22, como otras sulfonamidas, se manifiesta como un ácido débil que se representa según:
HGlib « H+ + Glib-
A pH 3,20 el equilibrio se desplaza hacia la forma no ionizada (HGlib) logrando la supresión iónica, lo que implica que a menor pH aumente el tr al incrementarse el grado de afinidad con la fase estacionaria apolar. Al emplear un pH de 5,25, cercano al pKa del analito, se establece un equilibrio7 donde coexisten ambas especies: ionizada y no ionizada, que en su equilibrio dinámico, favorecen un menor tr; tomando en cuenta que no hay deformación ni desdoblamiento del pico, se estableció 5,25 como pH de trabajo.
La presencia de impurezas limita la aplicación de muchos otros métodos por CLAR; la elevada especificidad de este procedimiento permite una amplia aplicación en las condiciones descritas con pocas interferencias las que fueron identificadas como: ibuprofeno, indometacina, trifluoperazina HCl, tioridazina HCl e imipramina HCl con diferentes grados de afectación sobre la cuantificación de Glib, por lo que el sistema mostró una alta selectividad, con la proporción de componentes de la FM y el pH crítica para un óptimo resultado.
Los estudios de linealidad, precisión y exactitud cumplieron con los parámetros y criterios estadísticos establecidos,13 por lo que el método resultó validado completamente para su ejecución, y quedaron fijados el LD y LC en 10 y 40 ng/mL respectivamente, al sobrepasar la relación señal/ruido en 3 y 10 unidades respectivamente, lo que permite alcanzar en el caso de que sea necesario niveles muy bajos en la cuantificación con suficiente exactitud como muestran los CV determinados a estas concentraciones, situación altamente favorable por los bajos niveles de medición a los que se enfrenta el procedimiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Guide to cleaning validation in API plants, cleaning validation in active pharmaceutical ingredient manufacturing plants. Active Pharmaceutical Ingredients Comité 1999; September: 7-20. Available from: http://apic.cefic.org/pub/4CleaningVal9909.pdf
2. Venkatesh P. Simultaneous determination of six antidiabetic drugs: glibenclamide, glicazide, glipizide, pioglitazone, repaglinide and rosiglitazone: development of a novel CLAR method for use in the analysis of pharmaceutical formulations and its application to human plasma assay. Biomed Chromatography. 2006;20(10):1043-8.
3. Kumasaka K. Screening and quantitative analysis for sulphonylurea-type oral antidiabetic agents in adulterated health food using thin layer chromatography and high performance liquid chromatography. J Health Sci. 2005;51:453-60.
4. Carabias-Martínez R. Simultaneous determination of phenyl and sufonylureas in water by solid phase extraction and liquid chromatography with diode array or mass spectrometric detection. Analytica Chemica Acta. 2004;517:71-9.
5. Pelander A. Toxicological screening with formula based metabolite identification by liquid chromatography/time of flight mass spectrometry. Analytical Chemistry. 2003;75:5710-18.
6. Prabu LS, Suriyaprakash TNK. Cleaning validation and its importance in pharmaceutical industry. Pharma Times. 2010;42(7):21-5.
7. PDA Tecnical Report No 29: Points to consider for cleaning validation. International association for Pharmaceutical Science and technology, Draf, March, 1998. Available from: http://www.pharmanet.com.br/pdf/tr29-002.pdf
8. LeBlanc DA. Cleaning Validation Teccnologies. Available from: http://www.cleaningvalidation.com/
9. López Marzo AM, Pierre Marzo RA. Establecimiento del límite aceptable para el residuo de limpieza en los equipos de producción de la industria farmacéutica. Rev Cubana Farm 2005;39(3). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75152005000300010&lng=es&nrm=iso
10. Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme. Available from: http://www.picscheme.org/
11. Fourman GL, Mullen MV. Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations. Pharm Technol. 1993;17(4):54-60.
12. Quattrochi O, Abelaira S. Introducción a la HPLC. Buenos Aires: Artes Gráficas; 1992.
13. Harmonized guidelines for single-laboratory validation of methods of analysis. Pure Applied Chemistry. 2002;74:835-55.
Recibido: 17 de octubre de 2011.
Aprobado: 29 de noviembre de 2011.
Alen Nils Baeza Fonte. Instituto de Ciencia y Tecnología de Materiales. Laboratorio de Análisis Químicos (LAQ). Universidad de La Habana. Zapata y G s/n. La Habana, Cuba. Correo electrónico: alen@imre.oc.uh.cu


{kind=link}