










My SciELO
 Custom services
Custom servicesServices on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.29 no.3 La Habana Jul.-Sept. 2012
REVIEW
Current treatment of rheumatoid arthritis. Perspectives for the development of antigen-specific therapies
Tratamiento actual de la artritis reumatoide. Perspectivas para el desarrollo de las terapias antígeno-específicas
Ariana Barberá, Noraylis Lorenzo, María del Carmen Domínguez
División de Química Física, Dirección de Investigaciones Biomédicas, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave 31 e/158 y 190. Cubanacán, Playa. PO Box 6162. La Habana, CP 10 600, Cuba.
ABSTRACT
Rheumatoid arthritis is a degenerative disease characterized by chronic inflammation of peripheral joints. The first line of treatment involves the use of potent anti-inflammatory and immunosuppressive drugs, leading to an overall suppression of the immune system. However, these drugs do not induce sustained remission and their use can cause immunosuppression that leads to severe complications. Thus, there is a need for developing new therapeutic alternatives for the treatment of this disease. Antigen-specific therapies allow the elimination of pathogenic cells without affecting the immune system's ability to respond to infections. Within this approach heat stress proteins are promising candidates. Although progress has been made in the development of efficient antigen-specific therapies, the excellent results obtained in animal models have been difficult to translate to humans. The combined use of antigen-specific therapy with current drugs can be an attractive strategy in the near future to achieve the complete remission of the disease. Some of these combinations have already begun to be evaluated in animal models and in rheumatoid arthritis patients.
Keywords: rheumatoid arthritis, regulatory T cells, biological therapy, pro-inflammatory cytokine, combination therapy.
RESUMEN
La artritis reumatoide es una enfermedad degenerativa caracterizada por la inflamación crónica de las articulaciones periféricas. La primera línea de tratamiento de esta enfermedad implica el uso de potentes antinflamatorios y drogas que provocan una supresión global del sistema inmune. Sin embargo, estos fármacos no inducen una remisión sostenida, y su uso puede causar una inmunosupresión importante que puede conducir a complicaciones. Por ello es necesario el desarrollo de nuevas modalidades terapéuticas para esta enfermedad. Las terapias antígeno-específicas suprimen las células patogénicas, sin afectar la propiedad del sistema inmune de responder ante las infecciones. Las proteínas de estrés térmico son candidatas promisorias en esta modalidad de tratamiento. Aunque se ha avanzado en el desarrollo de terapias antígeno-específicas eficientes en modelos animales con excelentes resultados, ha sido difícil trasladarlas a los seres humanos. El uso combinado de las terapias antígeno-específicas con los fármacos actuales puede ser una estrategia muy atractiva en el futuro cercano para lograr la remisión completa de la enfermedad. Algunas de estas combinaciones de tratamiento ya han comenzado a evaluarse en modelos animales y en pacientes con artritis reumatoide.
Palabras clave: artritis reumatoide, células T reguladoras, terapia biológica, citocina proinflamatoria, terapia combinada.
INTRODUCTION
Rheumatoid arthritis (RA), a systemic autoimmune disease most prevalent among adults between the fourth and fifth decade of life, currently affects 1% of the world population. Its primary clinical manifestation is the inflammation of peripheral joints, usually following a symmetrical pattern and exhibiting a chronic and progressive evolution that is characterized by periods of activity and remissions [1]. This disease produces a considerable deterioration of the quality of life of affected patients, owing in no small part to its chronic nature and the tendency to cause irreversible damage in cartilaginous and bony structures of the joints.
Currently, most RA therapies approved by the Food and Drug Administration (FDA) work by indiscriminately inhibiting all inflammatory activity in the patient. However, it is widely acknowledged that disease prognosis can be significantly improved by early treatment with disease-modifying anti-rheumatic drugs (DMARDs) [2, 3]. These otherwise unrelated compounds are neither painkillers nor anti-inflammatory drugs, but their use reduces, in the long term, the activity and severity of the disorder. Some well-known examples are methotrexate (MTX), leflunomide, antimalarial drugs such as chloroquine and hydroxychloroquine, gold salts, D-penicillamine, sulfasalazine and cyclosporin A.
During the nineties, MTX became first-line therapy for RA, propelled by therapeutic successes when combined with other drugs and an acceptable toxicity profile at the dosages used for this indication [4, 5]. MTX, however, is not exactly a wonder drug, as complete remission is achieved in only a fraction of RA cases [6]. In addition, it should be pointed out that the non-selective inhibition of the immune response places the patient at increased risk from infectious diseases and cancer.
Biological therapy, an alternative for patients not responding to MTX or other DMARDs, constitutes the most recent addition to the anti-rheumatic arsenal. Using biologicals to treat this disorder affords the possibility of targeting, in a more specific fashion, only those components playing an important role in the pathogenesis of RA [7, 8].
MODULATION OF THE IMMUNE RESPONSE BY BLOCKING THE ACTION OF MOLECULES INVOLVED IN THE PATHOGENESIS OF RA
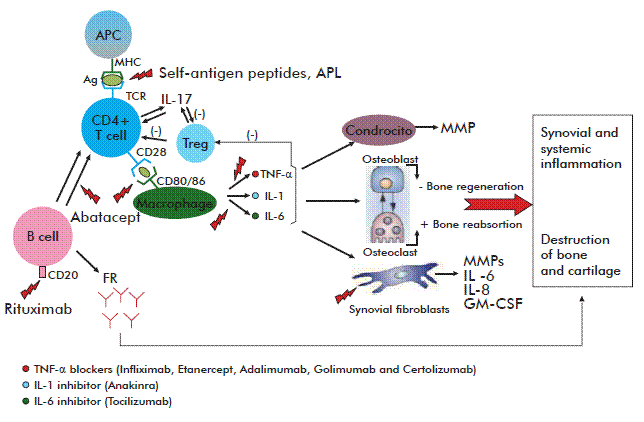
The last years have witnessed significant advances in the study and understanding of the mechanisms underlying the RA pathogenesis. It is known that the chronic joint inflammation produced by this disorder is induced by activated T cells infiltrating the synovial membrane. According to the most widely accepted hypothesis, damage is mediated through the recognition, by these cells, of a viral or bacterial molecule (or even a self-antigen). The recognition of this hypothetic antigen by CD49], triggers the differentiation of these lymphocytes into Th1 and Th17 phenotypes [10, 11]. Th1 and Th17 lymphocytes in turn secrete pro-inflammatory cytokines + T-cells, coupled with the stimulation of different cytokines [that then activate B and further T lymphocytes, macrophages and synovial cells, resulting in even higher levels of secreted cytokines, up-regulated adhesion molecules on cell surfaces and the migration of other inflammatory cells into the affected joint. Interleukin 1 (IL-1) and Tumor Necrosis Factor-alpha (TNF-α) are among the molecules whose secretion is up-regulated in the inflammatory environment produced by the activation of synovial cells and macrophages, and constitute the principal mediators of joint damage during the disease. These cytokines inhibit osteoblast-driven bone regeneration and stimulate reabsorption by osteoclasts, thereby contributing to the degeneration of bone tissue [9]. Fibroblasts attracted to the synovial membrane by TNF-α and IL-1 secrete, in turn, an array of molecules such as metalloproteases, prostaglandins, IL-6 and IL-8 which, acting in concert, increase the severity of joint damage. For example, metalloproteases play a role in the degeneration of the bone matrix; prostaglandins are pro-inflammatory molecules and IL-8 facilitates neutrophil chemotaxis and angiogenesis. The latter is especially important, in light of the hypoxic state and increased number of cells characterizing the articular space during this disorder. Activated synovial fibroblasts also produce the pro-inflammatory chemokines CCL5 and CXCL1, which actively recruit T and B lymphocytes as well as neutrophils into the synovial tissue. Likewise, they secrete IL-6, thus stimulating the production of acute phase reactants that worsen the inflammatory response. The resulting chronic inflammatory process leads to the proliferation of synovial tissue and the growth of a granular tissue denominated pannus, which exhibits the characteristics of a local multi-center tumor and eventually leads to the erosion of cartilage and bone [12].
As implied above, the term ‘biological agents’ or ‘biological therapy’ denotes complex protein molecules that can target in a very specific manner separate components of the pathogenic cascade leading to RA. These targets may range from cell surface molecules to cytokines or their receptors, also including adhesion or co-stimulation molecules involved in activation or effector mechanisms of the immune system. The figure represents the pathogenic mechanism of RA and the main points targeted by biological therapy.
There are a number of FDA-approved biologicals that can be used to block the main pro-inflammatory cytokines involved in the pathogenesis of RA.
TNF-α INHIBITORS
TNF-α is a cytokine that plays a crucial role in the pathogenesis of RA. This molecule, which binds specific receptors in the surface of cells from most human tissues, is found at high concentrations at the synovial membranes of RA patients. Initial reports described that blocking TNF-α in cultures of synovial tissue from inflamed joints resulted in reductions of the level of many pro-inflammatory mediators, such as IL-1, IL-6, IL-8 and granulocyte-macrophage colony stimulating factor [13]. Although reducing the level of pro-inflammatory molecules is essential for the efficacy of anti-TNF-α drugs, it goes without saying that pro-inflammatory cytokines are also essential for an effective immune response against microorganisms, and their reduction is the underlying cause of the severe adverse events sometimes associated with the use of these agents.
There are five commercially available TNF-α blockers approved for the treatment of RA: Etanercept® (fusion protein containing the soluble TNF-α receptor), Infliximab® (chimeric anti-TNF-α antibody), Adalimumab® (recombinant human anti-TNF-α antibody), Golimumab (humanized anti-TNF-α monoclonal antibody, mAb) and Certolizumab (PEGylated Fab fragment derived from a humanized mAb).
Currently, anti-TNF-α therapy constitutes the most successful treatment course for RA patients. The systemic suppression of TNF-α in this population has been associated, however, with increased susceptibility to infectious diseases, including in particular the reactivation of latent infections such as tuberculosis [14, 15], and a higher incidence of lymphomas, especially when anti-TNF-α drugs are administered in combination with MTX or corticosteroids [15, 16]. Paradoxically, despite the reduced severity of the signs and symptoms of RA offered by this treatment, it seems like blocking TNF-α for a prolonged period may increase the incidence of other autoimmune disorders such as multiple sclerosis and systemic lupus erythematosus [17, 18]. In addition, this therapy is effective only in 50% of RA cases [19].
IL-1 INHIBITOR
A single IL-1 inhibitor has been approved by the FDA for the treatment of RA: Anakinra®, an antagonist of the IL-1 receptor indicated for patients where the administration of anti-TNF-α agents has failed to produce a clinically meaningful improvement.
The efficacy of this drug, as estimated from early clinical trials, is not as high as that exhibited by TNF-α blockers. Lower efficacy, combined with the need for daily administration, explains why Anakinra® is less frequently used than TNF-α blockers in clinical practice [20].
IL-6 INHIBITOR
Tocilizumab® is a humanized mAb against the IL-6 receptor that has been shown to reduce the serum levels of vascular endothelial growth factor and protein C in RA patients, decreasing the severity of the signs and symptoms of the disease [21, 22]. The clinical efficacy and side effects associated with this inhibitor are comparable to those of anti-TNF-α drugs.
The efficacy in medical practice of biological agents that block pro-inflammatory cytokines is substantiated by a large number of clinical trials in patients suffering from RA and other autoimmune diseases [7, 8]. Unfortunately, none of these agents is able to achieve a complete remission of the disease. Such an outcome would require treatment for long periods, an unrealistic option in light of the severity of the side effects of these drugs.
Other therapeutic strategies have focused on modulating different elements of the adaptive immune response, ranging from antibodies blocking adhesion molecules that play an important role in lymphocyte traffic towards articular tissues to the depletion of T or B cells. Examples of the latter include attempts to delete only pathogenic cell populations (cells expressing OX-40) [23] or to deplete the antigens involved in T-cell signaling, such as the co-stimulatory molecules CD28, CD80 and CTLA-4 [24]. Some of these strategies have been shown to be effective in clinical trials, and have consequently been approved by the FDA for the treatment of RA.
INHIBITION OF CO-STIMULATION
Abatacept® is a fusion protein composed of the CTLA-4 molecule linked to the constant region of human IgG1. It represents the first example of the class of therapeutic drugs known as selective modulators of co-stimulation. This protein interacts with the CD80 and CD86 antigens at the surface of antigen-presenting cells (APCs), blocking their co-stimulatory interaction with CD28 and inhibiting, consequently, the activation of T lymphocytes. The efficacy of this approach was demonstrated in several clinical trials recruiting RA patients for which previous treatment with MTX had not been effective [25, 26].
B-LYMPHOCYTE DEPLETION
Depleting the B-lymphocyte population as a means to reduce or eliminate the cells producing pathogenic autoantibodies is an approach that has been previously tried for a wide range of autoimmune disorders. Rituximab®, a chimeric anti-CD20 mAb, is used in the context of RA. CD20 is a pan-B antigen whose expression is restricted to pre-B and mature B cells. It is absent in pluripotent stem cells, and is lost once B lymphocytes differentiate into plasma cells [27]. A number of clinical trials have shown Rituximab® to be an effective alternative for treating RA patients [28-30].
In general, the use of immunotherapy against this group of molecules represents a conceptual step forward, as it is aimed at modulating elements of the adaptive immune response rather than blocking an individual pathway (a cytokine, for instance). It must be noted, however, that this therapy also fails to produce a complete remission.
MODULATION OF THE ACTIVITY OF SYNOVIAL FIBROBLASTS
Different studies have revealed that synovial fibroblasts play an important role in the maintenance and persistence of inflammatory conditions at the articular microenvironment [12]. A number of recently developed therapeutic strategies target exactly this cell population, in addition to others that attempt to modulate fibroblast activation –TNF-α blockers, for instance– or to block its main products, using e.g. IL-6 inhibitors. There have also been attempts to target the cellular signaling pathways involved in the activation of these cells; Imatinib, for instance, is an inhibitor specific for a small family of tyrosine kinases that was first shown to modulate the proliferation of synovial fibroblasts in animal RA models [12]. This drug, approved by the FDA for the treatment of chronic myeloid leukemia and gastrointestinal tumors, inhibits several kinases involved in the pathogenesis of RA, such as the receptor for platelet-derived growth factor, which mediates fibroblast proliferation, the c-fms proto-oncogene, which participates in the activation and production of TNF-α in macrophages, and c-kit, which mediates secretion of TNF-α and IL-6. Imatinib was successful in an open study that recruited three patients with severe RA, recalcitrant to earlier treatments [31], and is currently under evaluation for other autoimmune and chronic infl ammatory disorders [32]. Work is also underway for developing treatments targeting phenotypic markers specifically associated with this cell population, or designed to modulate the epigenetic changes these cells undergo at the rheumatoid synovium [12].
A considerable number of studies have addressed the simultaneous use of DMARDs with biological drugs, as the clinical efficacy of this combination generally exceeds that of monotherapy. MTX with anti-TNF-α drugs constitutes the most successful and frequently employed combination, as it affords the patients a faster recovery of functional capacity and a slower radiologic progression [33-35].
In spite of their successes, the therapies mentioned above have failed to adequately address the challenge of restoring immune function to the point that the immune system can take over and maintain homeostasis after treatment discontinuation. The current situation, where DMARDs and biological drugs provide acceptable clinical control for most patients, has allowed the research community to shift its focus to strategies aimed at maintaining this disease-free state, and although the tools to achieve such a feat are not yet available, the general consensus points at the induction of immunological tolerance through the generation of antigen-specific regulatory T cells (Treg) as a possible solution [36].
PHENOTYPIC AND FUNCTIONAL CHARACTERISTICS OF REGULATORY T CELLS
The term ‘regulatory’ is currently employed to describe different populations of T cells that act over other cells of the immune system in an immunosuppressive fashion, both in vitro and in vivo. These populations play an important role in the maintenance of peripheral tolerance, the control of inflammatory responses against microorganisms, the control of intestinal homeostasis and the suppression of antitumoral immune responses [37, 38].
Regulatory T cells (Treg) can be traced back to two different populations, depending on their origin: natural Tregs and adaptive Tregs [39].
Natural Tregs are generated in the thymus, as a consequence of high-affinity interactions between their T-cell receptor (TCR) and major histocompatibility complex (MHC) molecules on the surface of stromal cells. Their TCR repertoire is every bit as diverse as that of conventional T cells [40]. Natural Tregs, which are released into peripheral compartments with an already stable and completely functional suppressor phenotype [41], account for close to 5% of the total number of CD4+ T lymphocytes [39]. Although they express constitutively the α chain of the IL-2 receptor (CD25), synthesis of this cytokine is barely detectable in Tregs, which therefore depend on exogenous IL-2 for their survival [42]. Other typical phenotypic markers of Tregs include CTLA-4 and the glucocorticoid-induced TNF receptor-related protein, although the one most frequently used for their identification is Foxp3, a transcription factor acting as the master switch of Treg ontogeny and maintenance. Despite recent studies demonstrating that this marker is transiently overexpressed in activated effector T lymphocytes and cannot, therefore, be considered exclusive for Tregs, Foxp3 is still used as an exclusive Treg marker for mouse cells [43].
Tregs exert their immunosuppressive effect through different mechanisms. Some depend on cell-cell contact and the action of granzymes A and B or perforins, which induce apoptosis in targeted effector T lymphocytes and other cell types such as monocytes and B lymphocytes [44, 45], or on direct contact with dendritic cells, affecting their maturation and functional capacity [46, 47]. Another immunosuppressive mechanism of Tregs is the secretion of soluble factors, including cytokines such as TGF-β [48], IL-10 [49] and IL-35 [50], as well as recently described immunosuppressive molecules such as carbon monoxide and galectin [51, 52].
In addition, and closely related to the addiction for exogenous IL-2 exhibited by peripheral Tregs, a mechanism has been described whereby these cells induce apoptosis on peripheral effector T-lymphocytes by depriving them of this cytokine [53].
Adaptive Tregs are generated at peripheral compartments from mature T cell populations, under very specific conditions of antigen exposure. Just like their natural counterparts, they have immunosuppressive properties and fulfill their function through mechanisms involving the secretion of cytokines and cell-cell contact [54]. Based on both genotypic and functional criteria, it has been possible to discern the existence of different subpopulations of adaptive Tregs: type 1 cells, which secrete IL-10, and Th3 cells, producing TGF-b. These cells are essential for maintaining homeostasis at the gastrointestinal tract [55, 56].
Treg CELLS AND RA
Treg cells are known to play a fundamental role in preventing the appearance of autoimmune disorders [57, 58]. A number of groups have therefore studied whether alterations in the number or activity of Tregs can contribute to the development of autoimmune diseases. In the specific case of RA, there are reports showing a higher frequency of T cells with a regulatory phenotype in synovial fluid [59-61]. Other studies have determined that RA patients exhibit higher frequencies of Treg cells in synovial fluid than in peripheral compartments, and have demonstrated that these patients have lower numbers of peripheral CD4+ CD25+ Tregs than healthy volunteers [62]. The higher numbers of Treg cells in the inflamed joint do not appear to be associated with disease severity or treatment efficacy [63], despite evidence linking the development of RA with increased frequencies of Treg cells at the synovium [64]. However, it has been demonstrated that the Treg cells of RA patients have suffered functional deterioration, since they are unable to inhibit the secretion of pro-inflammatory cytokines such as interferon gamma and TNF-α even though they are perfectly competent at suppressing the proliferation of autologous CD4+ CD25- T cells [65, 66]. This phenomenon might be caused by inhibitory effects mediated by some of the components of the environment of inflamed synovia, including cytokines such as TNF-α, IL-7, IL-15 and IL-6, whose concentration increases in RA patients [67, 68].
INDUCTION OF ANTIGEN-SPECIFIC Treg CELLS THROUGH THE MUCOSAL INDUCTION OF TOLERANCE USING SELF-ANTIGENS
Antigen-specific therapies for RA are aimed at eliminating only those T-cell clones that have escaped the control mechanisms of peripheral tolerance, contributing consequently to disease pathogenesis [69]. In theory, these therapies represent the best approach for manipulating the immune response in RA patients, as they spare the remaining cell populations of the immune system and must, therefore, be free of the side effects typically associated with general immunosuppression [70].
Antigen-specific tolerance can be induced by using the mucosal route to administer antigens typically found at the site of inflammation [71]. In many experimental models of autoimmune disease, the nasal or oral administration of a pathogenic self-antigen leads to considerable reductions of the severity of symptoms [72-75]. This phenomenon is mediated not only by the neutralization of antigen-specific T cells, but also by the induction of Tregs suppressing the effector functions of self-reactive T cells. The activation of Treg cells through the mucosal route is related to the presence of specialized APCs with tolerogenic properties at the gastrointestinal tract [76]. Antigen presentation at the gut takes place in the absence of co-stimulatory signals or accompanied by the expression of co-inhibitory signals in these APCs [77, 78]. Several studies, for instance, have shown that the ingestion of an antigen triggers the induction of TGF-β-secreting Th3 cells. Other groups have reported the induction of oral tolerance mediated by IL-10-secreting CD4+ CD25+ Treg cells [79-81].
There is experimental evidence suggesting that once activated, the suppressive activity of Tregs is not antigen-specific, targeting not only T cells stimulated by the same antigen but also activated CD4+ or CD8+ T-cell bystanders of unrelated specificities [82], probably through the secretion of inhibitory cytokines such as IL-10 and TGF-β. The fact that the induction of antigen-specific tolerance using Tregs was not restricted to a single antigenic specificity raised hopes concerning the efficacy of this alternative, which was expected to deliver a convincing performance in clinical settings [36].
However, despite the promising results obtained in animal models, so far this strategy has failed in clinical trials. According to available data, the oral administration of these antigens has been safe [83-87], but its clinical efficacy has been poor [83, 84] or restricted to small groups of patients [86]. It is known that the dose, frequency of administration and nature of the antigen itself are important variables that bear a tremendous influence in the outcome of mucosal tolerization. For instance, it has been shown that frequent administrations, as well as high doses, are not optimal for regulating the immune response in the intended manner [88]. Unfortunately, devising a clinical dose regime equivalent to those previously used with success in animal models is far from straightforward, and so far the use of suboptimal dosing schemes has made it impossible to realize the full potential of this approach.
It is also important to point out that in humans, the trigger antigens ultimately responsible for the appearance of RA are not as well defined as in animal models. The list of candidates spans both exogenous and autologous antigens, and there may be variations concerning the identity of trigger antigens from one individual to the next. To further compound matters, it is known that by the time the disorder becomes clinically evident, the autoimmune response has cascaded beyond the original triggers, affecting additional self-antigens in a phenomenon now known as epitope propagation [89]. Through this mechanism, the disorder may fall into a self-perpetuating cycle where the identity of the original trigger is ultimately irrelevant for clinical practice; another possible explanation as to why therapies aimed at inducing tolerance to chicken or bovine type II collagen, human cartilage glycoprotein-39, or the major constituents of articular cartilage, have failed to produce convincing results [90-92]. In order to break the cycle, therapy has to focus instead in the antigens currently fueling the amplification process, which must be overexpressed in situ and be recognized by the immune system. It must also be borne in mind that the molecular propagation mechanism mentioned above dictates that the selected antigen must also activate T cells regulating the pathogenic response to additional antigens involved in disease development at the inflammation site.
Heat shock proteins (HSPs) fulfill all of the above requirements. They are phylogenetically conserved, are highly immunogenic, and their expression is known to be ramped up during processes inducing cellular stress, such as inflammation [93]. HSPs are abundantly expressed at the inflammation site during RA [94, 95], where they –and derived peptides– are recognized as danger signals that can therefore trigger a strong pro-inflammatory response [94, 96, 97]. Although this type of response normally contributes to the elimination of nearby pathogens, the droves of endogenous HSP-derived peptides now present at the site thanks to cellular stress mechanisms may also become a new target for the immune system, thereby triggering a self-perpetuating inflammation cycle and amplifying the autoimmune process. One important property of these proteins, however, is that some HSP-derived self-peptides are able to induce regulatory T-cells [98], perhaps due to earlier contacts with exogenous HSPs throughout the life of the individual caused by previous immunizations or during interactions with food antigens [99].
In many ways, therefore, HSPs constitute ideal candidates for the induction of tolerance during autoimmune diseases, as they are able to stimulate a regulatory T-cell response and are abundantly represented at inflammation sites. Studies with cells isolated from patients affected with juvenile idiopathic arthritis, for example, have suggested that HSPs normally participate in the control of inflammation, where they are tasked with the induction of a regulatory response [100-103].
ANTIGEN-SPECIFIC IMMUNOMODULATION WITH HSP PEPTIDES
A protective role in experimental arthritis models has been demonstrated for numerous members of the HSP family [74, 104], some of which have even been evaluated in clinical trials. The first evidences suggesting that the 60 kDa HSP (HSP60) might be effective in the treatment of human arthritis were obtained during a study with OM-89, an Escherichia coli extract. Placebo-controlled multi-center clinical trials demonstrated that the extract was able to decrease the severity of the symptoms with few adverse events [105, 106]. The composition of OM-89 was characterized in later studies, which revealed the presence of HSP [107] inducing a specific T-cell response upon oral administration in animal models [108]. HSPs are currently considered responsible for the therapeutic effect of OM-89 in RA, a concept that was proven by later trials such as that of Prakken et al., who induced tolerance in RA patients by oral immunization with peptide dnaJP1 from the E. coli HSP40 [109]. This peptide is similar to the corresponding peptide of the human HSP40 homologue [94], and contains a QKRAA motif that has been associated with RA. A pilot Phase I trial with this peptide, recruiting 15 RA patients for a 6-month long treatment, found that it was able to shift the pro-inflammatory phenotype of peptide-specific T-cells to a regulatory phenotype, increasing the production of anti-inflammatory cytokines such as IL-4 and IL-10 while decreasing the levels of TNF-α and interferon gamma (INF-γ). Later placebo-controlled Phase II trials involving 160 patients with active RA showed that treatment with peptide dnaJP1 was safe, well tolerated, and reduced the levels of TNF-α production upon further stimulation with the peptide. In addition, the trial detected a statistically significant difference between the treated and placebo arms regarding the American College of Rheumatology 20 and 50% improvement criteria (ACR20 and ACR50, respectively), suggesting that the treatment produced a clinically significant outcome [110]
A recent Phase II clinical trial recruiting type 1 diabetes mellitus patients provided evidence of immunological efficacy for a different peptide derived from HSP60, denominated DiaPep277TM. It was later demonstrated that DiaPep277TM. The treatment was well tolerated, preserved the function of pancreatic β cells and decreased the demand for exogenous insulin when compared to the placebo group [111]. It was later demonstrated that DiaPep277TM activates human CD4+ CD25+ Tregs [98].
Summarizing, the results from clinical trials performed with RA and type 1 diabetes mellitus patients evidence that HSP-derived peptides can be used to produce a phenotypic pro-inflammatory to tolerogenic shift in pathogenic T-cell clones, providing clinically relevant benefits to the patient without the need for general immunosuppression.
In addition to peptides containing self-antigen epitopes for the induction of peripheral tolerance, this therapeutic concept has also been evaluated in experimental models for numerous autoimmune disorders using altered peptide ligands (APLs).
ANTIGEN-SPECIFIC IMMUNOMODULATION USING APLs
APLs are analogues of the immunogenic peptides from which they are derived. They usually bear one or several substitutions at positions essential for their interaction with TCR or MHC molecules, thereby interfering or modifying the intracellular signaling cascade that leads to the activation of T cells [112].
Conceptually, it is possible to design APLs with similar properties to the original immunogenic peptide (agonists) that provide stronger T-cell responses to specific antigens. Such an effect would be advantageous in pathological settings such as those of infectious or neoplastic diseases. On the other hand, it is also possible to design peptides that antagonize the original immunogenic peptide (antagonists or partial agonists), which may be useful in the context of an autoimmune disorder. APLs classified as null are those that can be presented by the APCs, but cause no effect on T lymphocytes [112].
Efforts to dissect the mechanism of action of APLs have centered on the biochemical events of T-cell activation. Binding of an agonist ligand to the TCR usually leads to the recruitment of kinases and the complete phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) in all CD3 chains, activating the effector functions of the T cell. In certain circumstances, however, binding of the MHC-APL complex to the target TCR may produce altered phosphorylation patterns and modulate tyrosine kinase activity, thus generating changes in the response of these cells [113].
There is ample published evidence on the capacity of APLs to modulate the immune response in experimental models of autoimmune disorders. It has been shown that these peptides can induce anergy or apoptosis in activated T lymphocytes through the expression of FasL and TNF-α [114, 115]. Also, it has been proposed that APLs can act in vivo as mediators of active suppression through the induction of Tregs secreting suppressive cytokines [116, 117]. In addition, there are examples evidencing that APLs may have beneficial effects through a combination of tolerogenic mechanisms [118].
One prominent example is that of Copaxone®, classified as an APL for its mechanism of action, which has been successfully used since the last decade for the treatment of multiple sclerosis. Copaxone® is a random sequence polymer synthesized from the aminoacids L-Ala, L-Tyr, L-Lys and L-Glu, which can be presented by numerous MHC class II molecules and acts as an antagonist of the immunodominant epitope of myelin basic protein. Binding of this compound to the TCR of autoreactive T cells specific for this immunodominant epitope induces tolerogenic effects in the latter [119]. In addition, it has been shown that the compound changes cytokine profi les from proinflammatory to regulatory, an effect involved in the mechanism of Treg-mediated active suppression that may explain the therapeutic effi cacy of this drug [120, 121].
As mentioned above, HSPs are prime candidates for the induction of antigen-specific tolerance in autoimmune disorders. Some research groups have designed APLs for different HSP60 epitopes, intended to expand Treg clones involved in active suppression. Currently, these APLs are still undergoing preclinical research.
One example is that of the immunodominant epitope of human HSP60 (180-188), used by Prakken et al. to design an APL that produced, upon intranasal administration, a prophylactic and therapeutic effect vastly superior to that of the native peptide in a model of adjuvant-induced arthritis. The designed APL bound the MHC of rats (RT1B1) with higher affinity, inducing IL-10-secreting Tregs that controlled disease progression [73].
Domínguez et al. used bioinformatics tools to select a T-cell epitope-rich region of HSP60 [122]. The selected peptide was modified at one of its predicted anchor residues for class II MHC molecules, a change that enabled its presentation in the context of several of the class II MHC molecules included in the study. Unlike its wild-type counterpart, the APL was able to expand CD4+ T-cell clones with a regulatory phenotype in Balb/C mice and in ex vivo assays using mononuclear cells isolated from RA patients. The peptide, in addition, exhibited a potent clinical and histopathological effect in two animal models of RA, which correlated with decreased levels of TNF-α [123].
This APL induces the apoptosis of peripheral blood lymphocytes with a memory phenotype in RA patients (unpublished observations). It is worth noting that this lymphocyte population has repeatedly been shown to be involved in the perpetuation of autoimmune inflammatory processes [124]. The biological effects of this APL, therefore, may arise from a combination of tolerogenic mechanisms that can be useful for the treatment of early-onset RA cases.
APLs, in general, have outperformed their originating peptides during testing in animal models. The first clinical trials of this concept, unfortunately, have told a different story. Two trials were held in 2000 for multiple sclerosis patients, who received two APLs derived from a T-cell epitope of myelin basic protein (residues 83-99), bearing substitution at positions heavily involved in peptide-TCR contacts. Both studies had to be interrupted after the appearance of systemic hypersensitivity reactions and the worsening of disease symptoms in three patients [125, 126]; effects that correlated with the expansion of antigen-specific Th1 cells during treatment. The researchers conducting the trial concluded that these effects were caused by the immunization protocol selected for both trials, which contemplated large (50 mg) subcutaneous doses favoring the expansion of T-cell clones with a Th1 phenotype; a conclusion further supported by the fact that the treatment was better tolerated by volunteers receiving the lowest dose (5 mg) in both studies. As a matter of fact, decreases in the number and volume of the lesions were detected in the immunized volunteers 4 months after treatment interruption.
These two failures prompted a deeper analysis of the molecular mechanisms that can be leveraged to induce a state of tolerance through the use of APL. Tentative courses of action examined included further modifications to APL design, dosing schemes and administration routes, in an effort to minimize the potential risk of activating pathogenic T-cell clones. One of the recommendations arising from this analysis was to administer APLs at the lowest dosage still producing a meaningful biological effect, so as to facilitate the expansion of APL-specific T cells with a regulatory phenotype while minimizing the chances of cross-activating pathogenic T-cell clones [112]. Another recommendation was to apply antigen-specific therapies only to patients where the onset of autoimmune disorder is relatively recent, in an attempt to intervene while the inflammatory process has not yet entered a self-perpetuating cycle and, therefore, reactivity to self-antigens has not become widespread yet.
In year 2006 the results of a clinical trial evaluating an APL derived from human insulin that recruited recent-onset type 1 diabetes mellitus patients was published. The trial employed subcutaneous doses of 0.1, 1 and 5 mg. It showed that the treatment was safe and well tolerated, and demonstrated a shift of the pathogenic Th1 phenotype to a regulatory phenotype [127].
Effi cacy considerations aside, it is unrealistic to expect that a single immunotherapeutic APL can be used for every individual. For an APL to work, it must be bound by some of the MHC molecules of the patient; otherwise, it cannot be presented by APCs and will not, therefore, be able to modulate the activity of Tcells. For example, some researchers have designed APLs derived from HCgp39 and peptide 256-276 of type II collagen with a high affi nity for allele MHCDR4 (DRB1*0401), which is overrepresented among RA patients [128, 129]. Regardless, there will inevitably be a group of patients where this particular class II MHC allele is absent, thereby limiting the immunotherapeutic usefulness of this particular peptide. The heterogeneity of TCR/MHC/peptide interactions is another factor that must be accounted for, as a single APL will not always trigger an immunosuppressive event when presented in the context of different TCR/MHC combinations, and some T cells specific for its cognate antigen will therefore escape suppression. It is therefore important for the APL to induce Tregs that not only suppress the response against their cognate self-antigen, but also against other relevant self-antigens present at the site of infl ammation. In the particular case of autoimmune disorders such as RA, where the identity of the pathogenic antigen is unknown, inducing Treg cells with these characteristics would be essential, and using for this purpose Hsp-derived peptides that not only participate in the pathogenic process, but –unlike type II collagen or HCgp39– constitute natural regulators of the immune response, can make the difference between success and failure [130].
It must be underscored, though, that the etiology and pathology of RA are complex, and achieving remission for this disorder will inevitably require the application of several complementing strategies. The previous or concomitant inclusion of antigen-specific therapies within established treatments using drugs already approved for this disease is currently being examined, with promising results.
ANTIGEN-SPECIFIC TOLERIZATION AS A COMPLEMENT FOR COMBINATION THERAPY
As discussed above, a pro-inflammatory environment is one of the hallmarks of active RA. Most studies indicate that Tregs in RA patients are not deficient, but rather that their functionality has been compromised by this environment [131]. It then follows that it must be possible to increase the efficiency of antigen-specific tolerization by simply inhibiting inflammation. An initial anti-inflammatory therapy would help to neutralize the action of pro-inflammatory cytokines and/or T cells at the inflammation site, clearing the field for subsequent antigenic tolerization strategies. According to previous reports, TNF-α blockers may serve this purpose, as they have been shown to restore the number and function of Tregs in RA patients [132] and may contribute to a shift towards a regulatory cytokine profile in monocytes and T-cells of RA patients [133].
Such was the rationale of a study combining the induction of antigen-specific Tregs with anti-TNF-α drugs in a rat model of adjuvant-induced arthritis. While the administration to separate groups of an thritogenic HSP60 peptide (p180-188) through the nasal route or a single-dose course of Etanercept® failed to produce a signifi cant reduction of the clinical signs of the disease, a combined schedule consisting on the administration of a single dose of Etanercept® before the induction of mucosal tolerance with the HSP60 peptide did produce a signifi cant clinical and histopathological improvement as well as a shift in the cytokine profile towards a regulatory phenotype [134]. Importantly, the results of the therapeutic combination were similar to those of a full three-dose course of Etanercept®, implying that one possible benefit of the combination would be a signifi cant reduction in the number of doses of anti-cytokine therapy, together with its associated side effects.
There is also evidence suggesting that clinical efficacy improves when the induction of Tregs is combined with anti-inflammatory treatments. In the clinical trial described earlier where the dnaJP1 peptide was administered to RA patients, the clinical effect was more noticeable in the group that also received hydroxychloroquine [110]. The latter drug decreases the levels of TNF-α and IL-6, in addition to blocking protein processing at the APC [135, 136], creating a situation where antigen presentation is diminished and therefore amplifying the impact of peptide dnaJP1, which does not need to be degraded in order to exert its effect on the regulation of the immune system. Taken as a whole, the available data suggest that combining the induction of Tregs with anti-inflammatory treatments may provide considerable clinical benefit during RA therapy.
CONCLUSIONS
RA is a severe disorder that considerably degrades the quality of life of affected patients and shortens their lifespan. Current available therapeutic alternatives are unsatisfactory, as they center predominantly on managing pain, reducing inflammation and delaying the progressive deterioration of the joints that characterizes this disease. Recognizing early-onset RA as a medical emergency has decreased the time at which treatment starts, enabling physicians to deal with the disorder from earlier stages with disease-modifying anti-rheumatic drugs and biologicals. Despite excellent results in the patient group that does respond to these therapies, these drugs cause generalized immunosuppression and, therefore, severe adverse events; in addition, they are unable to provide a sustained state of remission and must therefore be administered uninterruptedly. These obstacles may, in theory, be surmounted by manipulating the immune system in a more specific manner; a therapeutic modality for which the Heat Shock Proteins (HSP) are excellent candidates. These proteins are able to induce Treg cells specific for antigens of the inflammation site, thus avoiding the generalized immunosuppression of more traditional therapies. HSP treatment has been shown in clinical trials to be safe and capable of providing significant clinical improvement. Moreover, given that most studies indicate that Treg cells in RA patients are not deficient, but functionally compromised by the pro-inflammatory environment of the arthritic joint, the efficacy of HSP-based therapies can be enhanced simply by inhibiting inflammation with currently available drugs, an application for which the latter can be administered at dosages much lower than those used in purely anti-inflammatory treatments. This combination would therefore improve considerably the quality of life of affected patients, as it would significantly moderate the side effects associated with anti-inflammatory drugs and reduce treatment costs. In a best-case scenario, antigen-specific therapy might become the only long-term treatment required for maintaining the disorder under control. Inducing antigen-specific Tregs would, therefore, provide a huge boost to the safety and efficacy of therapies currently in use for RA patients.
REFERENCES
1. Pratt AG, Isaacs JD, Mattey DL. Current concepts in the pathogenesis of early rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2009;23(1):37-48.
2. Anderson JJ, Wells G, Verhoeven AC, Felson DT. Factors predicting response to treatment in rheumatoid arthritis: the importance of disease duration. Arthritis Rheum. 2000;43(1):22-9.
3. Nell VP, Machold KP, Eberl G, Stamm TA, Uffmann M, Smolen JS. Benefit of very early referral and very early therapy with disease-modifying anti-rheumatic drugs in patients with early rheumatoid arthritis. Rheumatology (Oxford). 2004;43(7):906-14.
4. Kinder AJ, Hassell AB, Brand J, Brownfield A, Grove M, Shadforth MF. The treatment of inflammatory arthritis with methotrexate in clinical practice: treatment duration and incidence of adverse drug reactions. Rheumatology (Oxford). 2005;44(1):61-6.
5. Klareskog L, van der Heijde D, de Jager JP, Gough A, Kalden J, Malaise M, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet. 2004;363(9410):675-81.
6. van Vollenhoven RF. Treatment of rheumatoid arthritis: state of the art 2009. Nat Rev Rheumatol. 2009;5(10):531-41.
7. Goldblatt F, Isenberg DA. New therapies for rheumatoid arthritis. Clin Exp Immunol. 2005;140(2):195-204.
8. Castro-Rueda H, Kavanaugh A. Biologic therapy for early rheumatoid arthritis: the latest evidence. Curr Opin Rheumatol. 2008;20(3):314-9.
9. Hoffmann M, Hayer S, Steiner G. Immmunopathogenesis of rheumatoid arthritis; induction of arthritogenic autoimmune responses by proinflammatory stimuli. Ann N Y Acad Sci. 2009;1173:391-400.
10. Boissier MC, Assier E, Falgarone G, Bessis N. Shifting the imbalance from Th1/Th2 to Th17/treg: the changing rheumatoid arthritis paradigm. Joint Bone Spine. 2008;75(4):373-5.
11. Gaffen SL. The role of interleukin-17 in the pathogenesis of rheumatoid arthritis. Curr Rheumatol Rep. 2009;11(5):365-70.
12. Juarez M, Filer A, Buckley C. Fibroblasts as therapeutic targets in rheumatoid arthritis and cancer. Swiss Med Wkly. 2012;142:50.
13. Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2(5):364-71.
14. Caspi RR. Immunotherapy of autoimmunity and cancer: the penalty for success. Nat Rev Immunol. 2008;8(12):970-6.
15. Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295(19):2275-85.
16. Feldmann M, Steinman L. Design of effective immunotherapy for human autoimmunity. Nature. 2005;435(7042):612-9.
17. De Bandt M. Lessons for lupus from tumour necrosis factor blockade. Lupus. 2006;15(11):762-7.
18. Lin J, Ziring D, Desai S, Kim S, Wong M, Korin Y, et al. TNFalpha blockade in human diseases: an overview of efficacy and safety. Clin Immunol. 2008;126(1):13-30.
19. Welsing PM, Severens JL, Hartman M, van Riel PL, Laan RF. Modeling the 5-year cost effectiveness of treatment strategies including tumor necrosis factor-blocking agents and leflunomide for treating rheumatoid arthritis in the Netherlands. Arthritis Rheum. 2004;51(6):964-73.
20. Jiang Y, Genant HK, Watt I, Cobby M, Bresnihan B, Aitchison R, et al. A multicenter, double-blind, dose-ranging, randomized, placebo-controlled study of recombinant human interleukin-1 receptor antagonist in patients with rheumatoid arthritis: radiologic progression and correlation of Genant and Larsen scores. Arthritis Rheum. 2000;43(5):1001-9.
21. Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67(11):1516-23.
22. Maini RN, Taylor PC, Szechinski J, Pavelka K, Broll J, Balint G, et al. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006;54(9):2817-29.
23. Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol. 2004;4(6):420-31.
24. Buch MH, Vital EM, Emery P. Abatacept in the treatment of rheumatoid arthritis. Arthritis Res Ther. 2008;10 Suppl 1:S5.
25. Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N Engl J Med. 2005;353(11):1114-23.
26. Schiff M, Keiserman M, Codding C, Songcharoen S, Berman A, Nayiager S, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi-centre, randomised, double-blind, placebo-controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis. 2008;67(8):1096-103.
27. Edwards JC, Leandro MJ, Cambridge G. B-lymphocyte depletion therapy in rheumatoid arthritis and other autoimmune disorders. Biochem Soc Trans. 2002;30(4):824-8.
28. Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54(9):2793-806.
29. Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54(5):1390-400.
30. Keystone E, Fleischmann R, Emery P, Furst DE, van Vollenhoven R, Bathon J, et al. Safety and efficacy of additional courses of rituximab in patients with active rheumatoid arthritis: an open-label extension analysis. Arthritis Rheum. 2007;56(12):3896-908.
31. Eklund KK, Joensuu H. Treatment of rheumatoid arthritis with imatinib mesylate: clinical improvement in three refractory cases. Ann Med. 2003;35(5):362-7.
32. Paniagua RT, Robinson WH. Imatinib for the treatment of rheumatic diseases. Nat Clin Pract Rheumatol. 2007;3(4):190-1.
33. Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54(1):26-37.
34. St Clair EW, van der Heijde DM, Smolen JS, Maini RN, Bathon JM, Emery P, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum. 2004;50(11):3432-43.
35. van der Heijde D, Klareskog L, Rodriguez-Valverde V, Codreanu C, Bolosiu H, Melo-Gomes J, et al. Comparison of etanercept and methotrexate, alone and combined, in the treatment of rheumatoid arthritis: two-year clinical and radiographic results from the TEMPO study, a double-blind, randomized trial. Arthritis Rheum. 2006;54(4):1063-74.
36. van de Ven A, Prakken B, Albani S. Immunological tolerance in the therapy of rheumatoid arthritis. Discov Med. 2007;7(37):46-50.
37. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775-87.
38. Wraith DC, Nicolson KS, Whitley NT. Regulatory CD4+ T cells and the control of autoimmune disease. Curr Opin Immunol. 2004;16(6):695-701.
39. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10(7):490-500.
40. Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006;25(2):249-59.
41. Picca CC, Larkin J, 3rd, Boesteanu A, Lerman MA, Rankin AL, Caton AJ. Role of TCR specificity in CD4+ CD25+ regulatory T-cell selection. Immunol Rev. 2006;212:74-85.
42. Malek TR, Bayer AL. Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol. 2004;4(9):665-74.
43. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19(4):345-54.
44. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27(4):635-46.
45. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174(4):1783-6.
46. Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology. 2006;118(2):240-9.
47. von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol. 2005;6(4):338-44.
48. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201(7):1061-7.
49. Suri-Payer E, Cantor H. Differential cytokine requirements for regulation of autoimmune gastritis and colitis by CD4(+)CD25(+) T cells. J Autoimmun. 2001;16(2):115-23.
50. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450(7169):566-9.
51. Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood. 2007;109(5):2058-65.
52. Lee SS, Gao W, Mazzola S, Thomas MN, Csizmadia E, Otterbein LE, et al. Heme oxygenase-1, carbon monoxide, and bilirubin induce tolerance in recipients toward islet allografts by modulating T regulatory cells. FASEB J. 2007;21(13):3450-7.
53. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8(12):1353-62.
54. Verbsky JW. Therapeutic use of T regulatory cells. Curr Opin Rheumatol. 2007;19(3):252-8.
55. Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30(5):626-35.
56. Levings MK, Roncarolo MG. Phenotypic and functional differences between human CD4+CD25+ and type 1 regulatory T cells. Curr Top Microbiol Immunol. 2005;293:303-26.
57. Oh S, Rankin AL, Caton AJ. CD4+CD25+ regulatory T cells in autoimmune arthritis. Immunol Rev. 2010;233(1):97-111.
58. Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol. 2007;7(8):585-98.
59. Cao D, Borjesson O, Larsson P, Rudin A, Gunnarsson I, Klareskog L, et al. FOXP3 identifies regulatory CD25bright CD4+ T cells in rheumatic joints. Scand J Immunol. 2006;63(6):444-52.
60. Mottonen M, Heikkinen J, Mustonen L, Isomaki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol. 2005;140(2):360-7.
61. Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, et al. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp Med. 2005;201(11):1793-803.
62. van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. 2004;50(9):2775-85.
63. Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmstrom V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther. 2004;6(4):R335-46.
64. Nanki T, Hayashida K, El-Gabalawy HS, Suson S, Shi K, Girschick HJ, et al. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol. 2000;165(11):6590-8.
65. Cao D, Malmstrom V, Baecher-Allan C, Hafler D, Klareskog L, Trollmo C. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol. 2003;33(1):215-23.
66. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. 2004;200(3):277-85.
67. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299(5609):1033-6.
68. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood. 2006;108(1):253-61.
69. Chatenoud L. Immune therapies of autoimmune diseases: are we approaching a real cure? Curr Opin Immunol. 2006;18(6):710-7.
70. Wehrens EJ, van Wijk F, Roord ST, Albani S, Prakken BJ. Treating arthritis by immunomodulation: is there a role for regulatory T cells? Rheumatology (Oxford). 2010;49(9):1632-44.
71. Wauben MH. Immunological mechanisms involved in experimental peptide immunotherapy of T-cell-mediated diseases. Crit Rev Immunol. 2000;20(6):451-69.
72. Garcia G, Komagata Y, Slavin AJ, Maron R, Weiner HL. Suppression of collagen-induced arthritis by oral or nasal administration of type II collagen. J Autoimmun. 1999;13(3):315-24.
73. Prakken BJ, Roord S, van Kooten PJ, Wagenaar JP, van Eden W, Albani S, et al. Inhibition of adjuvant-induced arthritis by interleukin-10-driven regulatory cells induced via nasal administration of a peptide analog of an arthritis-related heat-shock protein 60 T cell epitope. Arthritis Rheum. 2002;46(7):1937-46.
74. Zonneveld-Huijssoon E, Roord ST, de Jager W, Klein M, Albani S, Anderton SM, et al. Bystander suppression of experimental arthritis by nasal administration of a heat shock protein peptide. Ann Rheum Dis. 2011;70(12):2199-206.
75. Pop SM, Wong CP, He Q, Wang Y, Wallet MA, Goudy KS, et al. The type and frequency of immunoregulatory CD4+ T-cells govern the efficacy of antigen-specific immunotherapy in nonobese diabetic mice. Diabetes. 2007;56(5):1395-402.
76. Min SY, Park KS, Cho ML, Kang JW, Cho YG, Hwang SY, et al. Antigen-induced, tolerogenic CD11c+,CD11b+ dendritic cells are abundant in Peyer's patches during the induction of oral tolerance to type II collagen and suppress experimental collagen-induced arthritis. Arthritis Rheum. 2006;54(3):887-98.
77. Mason KL, Huffnagle GB, Noverr MC, Kao JY. Overview of gut immunology. Adv Exp Med Biol. 2008;635:1-14.
78. Zhang Y, Chung Y, Bishop C, Daugherty B, Chute H, Holst P, et al. Regulation of T cell activation and tolerance by PDL2. Proc Natl Acad Sci USA. 2006;103(31):11695-700.
79. Hultkrantz S, Ostman S, Telemo E. Induction of antigen-specific regulatory T cells in the liver-draining celiac lymph node following oral antigen administration. Immunology. 2005;116(3):362-72.
80. Mizrahi M, Ilan Y. The gut mucosa as a site for induction of regulatory T-cells. Curr Pharm Des. 2009;15(11):1191-202.
81. Battaglia M, Gianfrani C, Gregori S, Roncarolo MG. IL-10-producing T regulatory type 1 cells and oral tolerance. Ann N Y Acad Sci. 2004;1029:142-53.
82. Weiner HL. Oral tolerance: immune mechanisms and the generation of Th3-type TGF-beta-secreting regulatory cells. Microbes Infect. 2001;3(11):947-54.
83. Barnett ML, Kremer JM, St Clair EW, Clegg DO, Furst D, Weisman M, et al. Treatment of rheumatoid arthritis with oral type II collagen. Results of a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 1998;41(2):290-7.
84. Choy EH, Scott DL, Kingsley GH, Thomas S, Murphy AG, Staines N, et al. Control of rheumatoid arthritis by oral tolerance. Arthritis Rheum. 2001;44(9):1993-7.
85. Myers LK, Higgins GC, Finkel TH, Reed AM, Thompson JW, Walton RC, et al. Juvenile arthritis and autoimmunity to type II collagen. Arthritis Rheum. 2001;44(8):1775-81.
86. Sieper J, Kary S, Sorensen H, Alten R, Eggens U, Huge W, et al. Oral type II collagen treatment in early rheumatoid arthritis. A double-blind, placebo-controlled, randomized trial. Arthritis Rheum. 1996;39(1):41-51.
87. Trentham DE, Dynesius-Trentham RA, Orav EJ, Combitchi D, Lorenzo C, Sewell KL, et al. Effects of oral administration of type II collagen on rheumatoid arthritis. Science. 1993;261(5129):1727-30.
88. Peakman M, von Herrath M. Antigen-specific immunotherapy for type 1 diabetes: maximizing the potential. Diabetes. 2010;59(9):2087-93.
89. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2(2):85-95.
90. Keystone EC. Abandoned therapies and unpublished trials in rheumatoid arthritis. Curr Opin Rheumatol. 2003;15(3):253-8.
91. Landewe RB, Houbiers JG, Van den Bosch F, in't Hout J, Verschueren PC, Meijerink JH, et al. Intranasal administration of recombinant human cartilage glycoprotein-39 as a treatment for rheumatoid arthritis: a phase II, multicentre, double-blind, randomised, placebo-controlled, parallel-group, dose-finding trial. Ann Rheum Dis. 2010;69(9):1655-9.
92. McKown KM, Carbone LD, Kaplan SB, Aelion JA, Lohr KM, Cremer MA, et al. Lack of efficacy of oral bovine type II collagen added to existing therapy in rheumatoid arthritis. Arthritis Rheum. 1999;42(6):1204-8.
93. van Eden W, van der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5(4):318-30.
94. Albani S, Keystone EC, Nelson JL, Ollier WE, La Cava A, Montemayor AC, et al. Positive selection in autoimmunity: abnormal immune responses to a bacterial dnaJ antigenic determinant in patients with early rheumatoid arthritis. Nat Med. 1995;1(5):448-52.
95. Boog CJ, de Graeff-Meeder ER, Lucassen MA, van der Zee R, Voorhorst-Ogink MM, van Kooten PJ, et al. Two monoclonal antibodies generated against human hsp60 show reactivity with synovial membranes of patients with juvenile chronic arthritis. J Exp Med. 1992;175(6):1805-10.
96. Albani S. Infection and molecular mimicry in autoimmune diseases of childhood. Clin Exp Rheumatol. 1994;12 Suppl 10:S35-41.
97. Albani S, Ravelli A, Massa M, De Benedetti F, Andree G, Roudier J, et al. Immune responses to the Escherichia coli dnaJ heat shock protein in juvenile rheumatoid arthritis and their correlation with disease activity. J Pediatr. 1994;124(4):561-5.
98. Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116(7):2022-32.
99. van Eden W, van der Zee R, Paul AG, Prakken BJ, Wendling U, Anderton SM, et al. Do heat shock proteins control the balance of T-cell regulation in inflammatory diseases? Immunol Today. 1998;19:303-7.
100. de Kleer IM, Kamphuis SM, Rijkers GT, Scholtens L, Gordon G, De Jager W, et al. The spontaneous remission of juvenile idiopathic arthritis is characterized by CD30+ T cells directed to human heat-shock protein 60 capable of producing the regulatory cytokine interleukin-10. Arthritis Rheum. 2003;48(7):2001-10.
101. Kamphuis S, Kuis W, de Jager W, Teklenburg G, Massa M, Gordon G, et al. Tolerogenic immune responses to novel T-cell epitopes from heat-shock protein 60 in juvenile idiopathic arthritis. Lancet. 2005;366(9479):50-6.
102. Massa M, Passalia M, Manzoni SM, Campanelli R, Ciardelli L, Yung GP, et al. Differential recognition of heat-shock protein dnaJ-derived epitopes by effector and Treg cells leads to modulation of inflammation in juvenile idiopathic arthritis. Arthritis Rheum. 2007;56(5):1648-57.
103. de Graeff-Meeder ER, van Eden W, Rijkers GT, Prakken BJ, Kuis W, Voorhorst-Ogink MM, et al. Juvenile chronic arthritis: T cell reactivity to human HSP60 in patients with a favorable course of arthritis. J Clin Invest. 1995;95(3):934-40.
104. Cobelens PM, Heijnen CJ, Nieuwenhuis EE, Kramer PP, van der Zee R, van Eden W, et al. Treatment of adjuvant-induced arthritis by oral administration of mycobacterial Hsp65 during disease. Arthritis Rheum. 2000;43(12):2694-702.
105. Rosenthal M, Bahous I, Ambrosini G. Longterm treatment of rheumatoid arthritis with OM-8980. A retrospective study. J Rheumatol. 1991;18(12):1790-3.
106. Vischer TL. Follow-up with OM-8980 after a double-blind study of OM-8980 and auranofin in rheumatoid arthritis. Clin Rheumatol. 1990;9(3):356-61.
107. Polla BS, Baladi S, Fuller K, Rook G. Presence of hsp65 in bacterial extracts (OM-89): a possible mediator of orally-induced tolerance? Experientia. 1995;51(8):775-9.
108. Bloemendal A, Van der Zee R, Rutten VP, van Kooten PJ, Farine JC, van Eden W. Experimental immunization with anti-rheumatic bacterial extract OM-89 induces T cell responses to heat shock protein (hsp)60 and hsp70; modulation of peripheral immunological tolerance as its possible mode of action in the treatment of rheumatoid arthritis (RA). Clin Exp Immunol. 1997;110(1):72-8.
109. Prakken BJ, Samodal R, Le TD, Giannoni F, Yung GP, Scavulli J, et al. Epitope-specific immunotherapy induces immune deviation of proinflammatory T cells in rheumatoid arthritis. Proc Natl Acad Sci USA. 2004;101(12):4228-33.
110. Koffeman EC, Genovese M, Amox D, Keogh E, Santana E, Matteson EL, et al. Epitope-specific immunotherapy of rheumatoid arthritis: clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial. Arthritis Rheum. 2009;60(11):3207-16.
111. Raz I, Elias D, Avron A, Tamir M, Metzger M, Cohen IR. Beta-cell function in new-onset type 1 diabetes and immunomodulation with a heat-shock protein peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet. 2001;358(9295):1749-53.
112. Bielekova B, Martin R. Antigen-specific immunomodulation via altered peptide ligands. J Mol Med (Berl). 2001;79(10):552-65.
113. Madrenas J, Wange RL, Wang JL, Isakov N, Samelson LE, Germain RN. Zeta phosphorylation without ZAP-70 activation induced by TCR antagonists or partial agonists. Science. 1995;267(5197):515-8.
114. Ben-David H, Sela M, Mozes E. Down-regulation of myasthenogenic T cell responses by a dual altered peptide ligand via CD4+CD25+-regulated events leading to apoptosis. Proc Natl Acad Sci USA. 2005;102(6):2028-33.
115. Katsara M, Deraos G, Tselios T, Matsoukas J, Apostolopoulos V. Design of novel cyclic altered peptide ligands of myelin basic protein MBP83-99 that modulate immune responses in SJL/J mice. J Med Chem. 2008;51(13):3971-8.
116. Ben-David H, Venkata Aruna B, Sela M, Mozes E. A dual altered peptide ligand inhibits myasthenia gravis associated responses by inducing phosphorylated extracellular-regulated kinase 1,2 that upregulates CD4+CD25+Foxp3+ cells. Scand J Immunol. 2007;65(6):567-76.
117. Zhao J, Li R, He J, Shi J, Long L, Li Z. Mucosal administration of an altered CII263-272 peptide inhibits collagen-induced arthritis by suppression of Th1/Th17 cells and expansion of regulatory T cells. Rheumatol Int. 2008;29(1):9-16.
118. Anderton SM. Peptide-based immunotherapy of autoimmunity: a path of puzzles, paradoxes and possibilities. Immunology. 2001;104(4):367-76.
119. Aharoni R, Teitelbaum D, Arnon R, Sela M. Copolymer 1 acts against the immunodominant epitope 82-100 of myelin basic protein by T cell receptor antagonism in addition to major histocompatibility complex blocking. Proc Natl Acad Sci USA. 1999;96(2):634-9.
120. Duda PW, Schmied MC, Cook SL, Krieger JI, Hafler DA. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest. 2000;105(7):967-76.
121. Neuhaus O, Farina C, Yassouridis A, Wiendl H, Then Bergh F, Dose T, et al. Multiple sclerosis: comparison of copolymer-1- reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc Natl Acad Sci USA. 2000;97(13):7452-7.
122. Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics. 2001;17(12):1236-7.
123. Dominguez M del C, Lorenzo N, Barbera A, Darrasse-Jeze G, Hernandez MV, Torres A, et al. An altered peptide ligand corresponding to a novel epitope from heat-shock protein 60 induces regulatory T cells and suppresses pathogenic response in an animal model of adjuvant-induced arthritis. Autoimmunity. 2011;44(6):471-82.
124. Szodoray P, Jellestad S, Nakken B, Brun JG, Jonsson R. Programmed cell death in rheumatoid arthritis peripheral blood T-cell subpopulations determined by laser scanning cytometry. Lab Invest. 2003;83(12):1839-48.
125. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6(10):1167-75.
126. Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, et al. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. 2000;6(10):1176-82.
127. Alleva DG, Maki RA, Putnam AL, Robinson JM, Kipnes MS, Dandona P, et al. Immunomodulation in type 1 diabetes by NBI-6024, an altered peptide ligand of the insulin B epitope. Scand J Immunol. 2006;63(1):59-69.
128. Boots AM, Hubers H, Kouwijzer M, den Hoed-van Zandbrink L, Westrek-Esselink BM, van Doorn C, et al. Identification of an altered peptide ligand based on the endogenously presented, rheumatoid arthritis-associated, human cartilage glycoprotein-39(263-275) epitope: an MHC anchor variant peptide for immune modulation. Arthritis Res Ther. 2007;9(4):R71.
129. Sakurai Y, Brand DD, Tang B, Rosloniec EF, Stuart JM, Kang AH, et al. Analog peptides of type II collagen can suppress arthritis in HLA-DR4 (DRB1*0401) transgenic mice. Arthritis Res Ther. 2006;8(5):R150.
130. Quintana FJ, Cohen IR. The HSP60 immune system network. Trends Immunol. 2011;32(2):89-95.
131. Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci USA. 2008;105(49):19396-401.
132. Toubi E, Kessel A, Mahmudov Z, Hallas K, Rozenbaum M, Rosner I. Increased spontaneous apoptosis of CD4+CD25+ T cells in patients with active rheumatoid arthritis is reduced by infliximab. Ann N Y Acad Sci. 2005;1051:506-14.
133. Schuerwegh AJ, Van Offel JF, Stevens WJ, Bridts CH, De Clerck LS. Influence of therapy with chimeric monoclonal tumour necrosis factor-alpha antibodies on intracellular cytokine profiles of T lymphocytes and monocytes in rheumatoid arthritis patients. Rheumatology (Oxford). 2003;42(4):541-8.
134. Roord ST, Zonneveld-Huijssoon E, Le T, Yung GP, Koffeman E, Ronaghy A, et al. Modulation of T cell function by combination of epitope specific and low dose anticytokine therapy controls autoimmune arthritis. PLoS One. 2006;1:e87.
135. Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. Effects of chloroquine on viral infections: an old drug against today's diseases? Lancet Infect Dis. 2003;3(11):722-7.
136. van den Borne BE, Dijkmans BA, de Rooij HH, le Cessie S, Verweij CL. Chloroquine and hydroxychloroquine equally affect tumor necrosis factor-alpha, interleukin 6, and interferon-gamma production by peripheral blood mononuclear cells. J Rheumatol. 1997;24(1):55-60.
Received in December, 2011.
Accepted for publication in March, 2012.
Ariana Barberá. División de Química Física, Dirección de Investigaciones Biomédicas, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, Habana 16, C.P. 11600, La Habana, Cuba. E-mail: ariana.barbera@cigb.edu.cu.

{kind=link}